微生物 研究
Background
背景
While a New York Subway station is bustling with swarms of businessmen, students, artists, and millions of other city-goers every day, its floors, railings, stairways, toilets, walls, kiosks, and benches are teeming with non-human life. The microbial ecosystem, or the complex web of relationships that microorganisms have with one another and with the environment, is omnipresent in shared public transportation spaces. The PathoMap, a 2013 project led by Cornell Professor Christopher Mason, was the first of an annual series of DNA collection projects at various locations around the world. A sampling from NYC subways found that only half of the DNA matched known organisms. This initial project confirmed that the urban microbiome was still a relatively unexplored field, virtually begging for researchers to seize the opportunity. The success of the project yielded the creation of the MetaSUB international consortium, and ever since, intensive studies have been carried out with microbial samples from urban locations around the globe. In addition to being a relatively new field of work, the applications of such projects are endless.
紐約地鐵站每天都擠滿了大批商人,學生,藝術家和數百萬其他城市居民,但它的地板,欄桿,樓梯,廁所,墻壁,信息亭和長椅上充斥著非人類的生活。 微生物生態系統,或微生物彼此之間以及與環境之間復雜的關系網,在共享的公共交通空間中無處不在。 由康奈爾大學教授克里斯托弗·梅森(Christopher Mason)領導的2013年PathoMap項目是全球各地每年一次的DNA收集項目的第一個系列。 從紐約地鐵的一個樣本中發現,只有一半的DNA與已知生物匹配。 該項目初始確認該城市的微生物仍然是一個相對未開發領域,幾乎乞求的研究人員抓住機會。 該項目的成功促成了MetaSUB國際財團的創立,從那時起,就對來自全球城市地區的微生物樣本進行了深入研究。 除了是一個相對較新的工作領域之外,此類項目的應用也無窮無盡。
But what happens after the samples are collected? As I learned this summer, there is no magical one-step formula that outputs clean, categorized, analyzed, and graphed data. I had the opportunity to work with MetaSUB microbial data and learn the painstaking yet satisfying process of biological data manipulation and visualization.
但是收集樣本后會怎樣? 正如我今年夏天了解到的那樣,沒有神奇的一步式公式可以輸出干凈,分類,分析和圖形化的數據。 我有機會處理MetaSUB微生物數據,并學習了艱苦而又令人滿意的生物數據處理和可視化過程。
Collecting & Cleaning the Data in Linux Bash Terminal
在Linux Bash Terminal中收集和清理數據
Before the analysis is run, swab samples are collected from specified locations, and DNA libraries are prepared for paired-end sequencing. Since both ends of the fragment are sequenced, this type of sequencing allows for more precise reading of the DNA, results in better alignment, and detects any rearrangements.
在運行分析之前,應從指定的位置收集拭子樣品,并準備DNA文庫用于雙末端測序。 由于片段的兩端均已測序,因此這種類型的測序可更精確地讀取DNA,更好的比對并檢測任何重排。
While the biological sampling yields a plethora of data, not all of it is relevant for analysis. Adapter sequences, low-quality bases, and human DNA were all extraneous data points in the set. For my project, the first goal was to clean and categorize this data in the Linux Bash Terminal to run visual analyses in R Studio.
盡管生物采樣產生了大量數據,但并非所有數據都與分析相關。 接頭序列,低質量堿基和人類DNA都是該集中的無關數據點。 對于我的項目,首要目標是在Linux Bash Terminal中清理和分類這些數據,以便在R Studio中運行可視化分析。
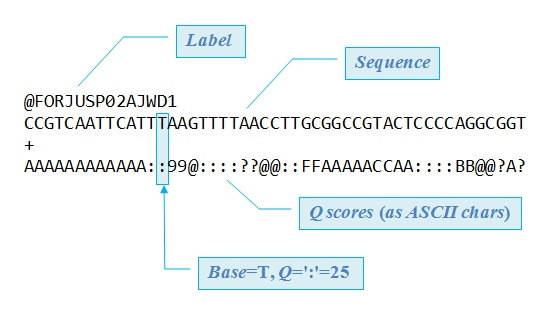
The Linux Bash Terminal is used for all reading and cleaning of the data. Read files are stored in a FASTQ format (as seen in Figure 1), and the following is the structure of this file type:
Linux Bash Terminal用于所有讀取和清除數據。 讀取文件以FASTQ格式存儲(如圖1所示),以下是該文件類型的結構:
- Line 1 of the file contains the identifier of the sequence which summarizes where the sequence of bases is found 文件的第1行包含序列的標識符,該標識符概括了在何處發現堿基序列
- Line 2 provides the actual raw letters of the nitrogenous base sequence 第2行提供了含氮堿基序列的實際原始字母
- Line 3 starts with the plus sign and may be followed by the sequence identifier again 第3行以加號開頭,并且可能再次由序列標識符跟隨
- Line 4 is the last line of the FASTQ file, and it contains the quality score of each base in the format of an ASCII symbol, each symbol corresponding to a Phred Quality (Q) score 第4行是FASTQ文件的最后一行,它包含ASCII符號格式的每個基準的質量得分,每個符號對應于Phred Quality(Q)得分
However, the raw files still have unwanted DNA fragments, including adapters (which are used to ligate the DNA molecules) and low-quality bases (which can be identified using the ASCII or Phred Quality scores). The Linux AdapterRemoval function is used to remove these adapter sequences and trim the data of low-quality bases, as seen in Figure 2.
但是,原始文件仍然具有不需要的DNA片段,包括銜接子(用于連接DNA分子)和低質量的堿基(可以使用ASCII或Phred Quality分數進行標識)。 Linux AdapterRemoval函數用于刪除這些適配器序列并修剪低質量基數的數據,如圖2所示。
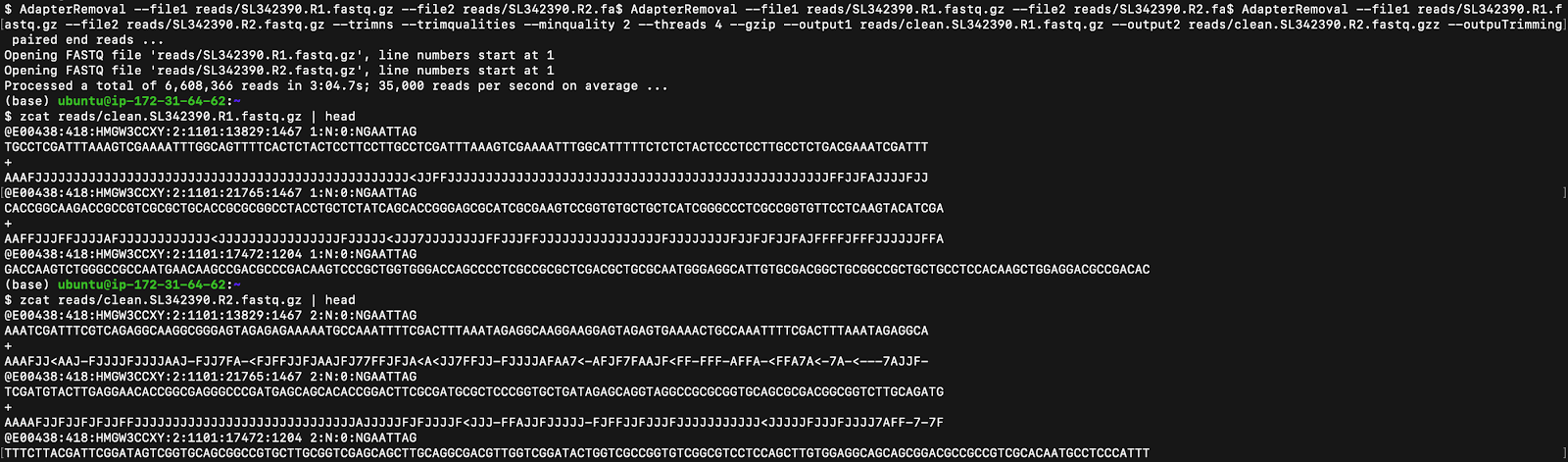
New FASTQ files are created from the trimmed reads after the sample reads are identified, the Adapter Removal function is used, the reads are filtered with a minimum quality score, and the files are unzipped as seen in the first line of code in Figure 2.
在識別樣本讀取后,使用修剪后的讀取創建新的FASTQ文件,使用適配器移除功能,以最低質量得分過濾讀取,并解壓縮文件,如圖2中第一行代碼所示。
Although these sequences have been removed, the reads are still not thoroughly cleaned. For this project, only microbial data is needed. However, DNA samples from a public transportation space will contain mostly (close to 99%) human DNA since the bacterial genome is more than 1600 times smaller than the human one. Fortunately, most of this human DNA has already been stripped in earlier stages of this project, though some may remain. The bowtie2 function in Linux must be used to completely rid the reads of human DNA.
盡管已刪除了這些序列,但仍未徹底清除讀段。 對于此項目,僅需要微生物數據。 但是,來自公共交通場所的DNA樣本將主要(接近99%)包含人類DNA,因為細菌基因組的大小是人類基因組的1600倍以上。 幸運的是,大多數人類DNA在該項目的早期階段已經被剝離,盡管可能還會保留一些。 Linux中的bowtie2函數必須用于完全清除人類DNA的讀碼。
The bowtie2 function works in a fairly intuitive way. Rather than going through each file and identifying whether or not there is human DNA (which would yield a terrible run time), the algorithm utilizes a human index reference genome. Many human reference genomes have been created for such purposes, and the most commonly used one is reference genome 38, which is used here. To provide an analogy for how the function works, the way I thought of this was like a book — to find a term or specific sentence in a book, it is far easier to check the index to see what page it is on rather than flipping through the full text. Likewise, suppose we provide bowtie with the information on the human reference genome. In that case, it will be much more efficient because the program will know where to go on the genome since the reference index narrows it down. From the function’s output in Figure 3, it can be seen that 0.95% of the genome aligned with human DNA, meaning that the reads were already thoroughly cleaned, but not yet entirely stripped as it is now. New files were created that had cleaned reads.
bowtie2函數以相當直觀的方式工作。 該算法不是遍歷每個文件并識別是否存在人類DNA(這將導致可怕的運行時間),而是利用人類索引參考基因組。 為此目的已經創建了許多人類參考基因組,最常用的是參考基因組38,在此使用。 為了給函數的工作方式提供一個類比,我想到的方式就像一本書—在書中查找一個術語或特定句子,檢查索引以查看它所在的頁面比翻頁要容易得多。通過全文。 同樣,假設我們向領結提供有關人類參考基因組的信息。 在這種情況下,它將更有效率,因為程序會知道基因組的位置,因為參考索引會縮小基因組的范圍。 從圖3的函數輸出中可以看出,基因組的0.95%與人類DNA對齊,這意味著讀取的片段已被徹底清洗,但尚未像現在一樣被完全剝離。 創建的新文件已清除讀取。
After cleaning the data of unwanted reads (which could have meant either adapter sequences, low-quality bases, or human DNA), it is essential to identify the amount of data we will be using. To count the total number of reads (and we will count them at each stage of the process up until now) the zcat function is used to calculate the total number of FASTQ file lines and the output is divided by four since all reads are spread across the four lines of each FASTQ file.
清除不需要的讀數(可能意味著銜接子序列,低質量的堿基或人類DNA)的數據后,確定我們將要使用的數據量至關重要。 要計算讀取的總數(到目前為止,我們將在過程的每個階段進行計數),zcat函數用于計算FASTQ文件行的總數,并且由于所有讀取分散在輸出中,因此輸出結果被四分每個FASTQ文件的四行。
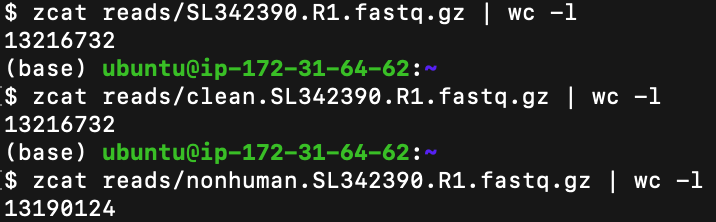
- Initial # of reads: 3,304,183 reads 初始讀取次數:3,304,183次讀取
- # of reads after AdapterRemoval: 3,304,183 適配器卸下后的讀取次數:3,304,183
- # of reads after Bowtie 2 Alignment: 3,297,531 Bowtie 2對齊后的讀取次數:3,297,531
Furthermore, to learn more about the data, the GC (guanine-cytosine percent) content is calculated using the qc-stats function, as seen in Figure 5.
此外,要了解有關數據的更多信息,請使用qc-stats函數計算GC(鳥嘌呤-胞嘧啶百分比)含量,如圖5所示。
The significance of the GC content is that there is a correlation between GC content and types of bacteria. Bacteria that might be specific to a particular biome may have comparatively lower or higher GC contents than those of other biomes. There is also a strong correlation between GC content and the stability of DNA, since G-C base pairs have one more hydrogen bond than A-T base pairs. Therefore, a higher GC content means higher required temperature for denaturing the DNA, and this information is useful for PCR amplification and identifying the source or environment of the bacteria. Next, using a program called Kraken, the taxonomically categorized data can be printed in the Linux Terminal, as seen in Figure 6.
GC含量的意義在于GC含量和細菌類型之間存在相關性。 特定于特定生物群落的細菌可能具有比其他生物群落相對較低或較高的GC含量。 GC含量與DNA穩定性之間也有很強的相關性,因為GC堿基對比AT堿基對具有更多的氫鍵。 因此,較高的GC含量意味著使DNA變性所需的較高溫度,并且此信息可用于PCR擴增和鑒定細菌的來源或環境。 接下來,使用稱為Kraken的程序,可以在Linux Terminal中打印分類分類的數據,如圖6所示。
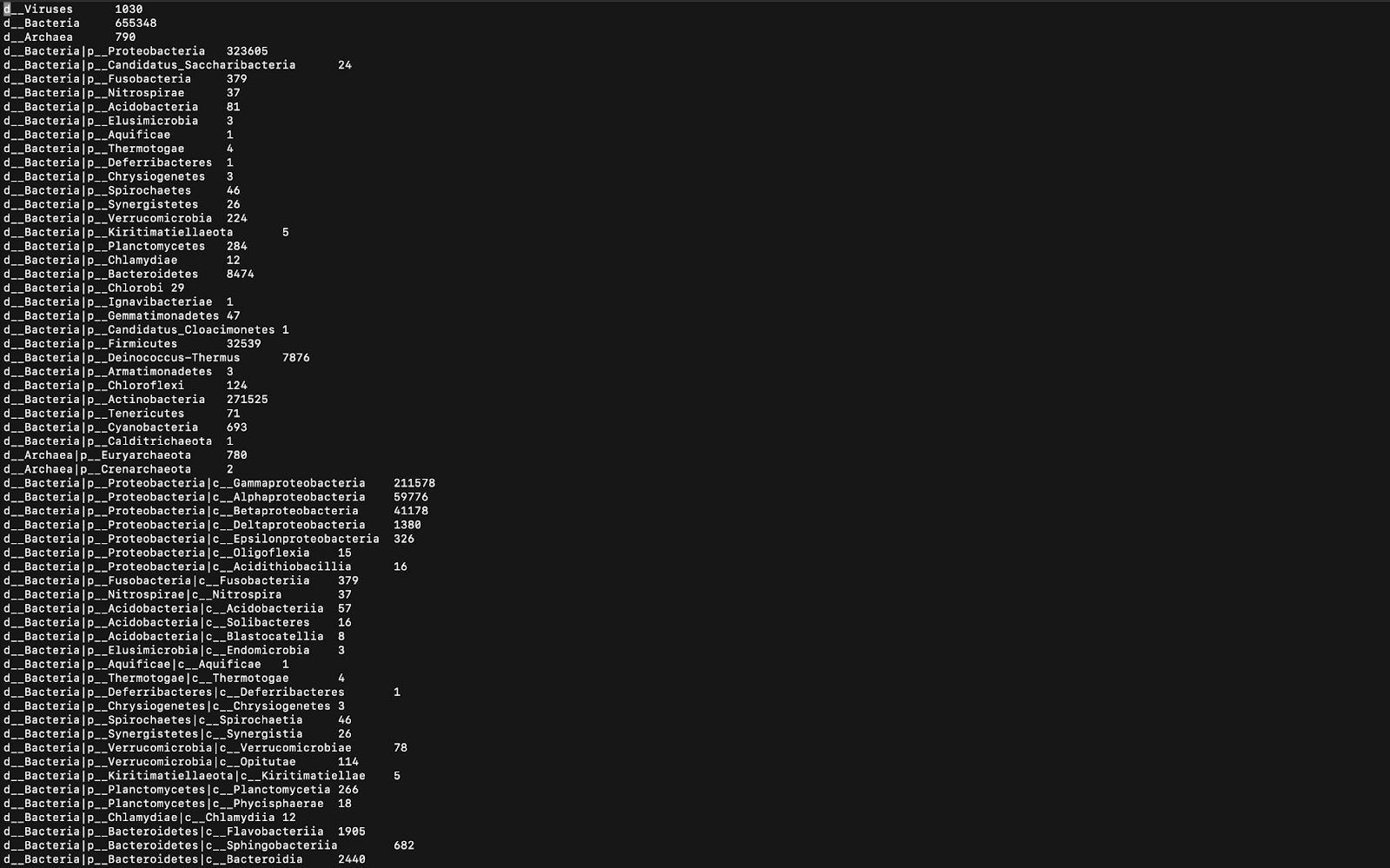
Kraken’s output is handy, as the actual classifications of the microbes can now be seen. In Figure 6, only a minimal number of the total classifications can be seen (that’s all that fit in the screenshot). The letters before the rankings are all abbreviations for Domain, Kingdom, Phylum, Class, Order, Family, Genus, and Species (the taxonomy order). The numbers at the end of each line represent the proportion of the corresponding species (or domain, phylum, etc. if the species was not identified) in the sample. The program is not able to identify many of the species because those are yet to be discovered.
Kraken的輸出非常方便,因為現在可以看到微生物的實際分類。 在圖6中,只能看到最小數量的總分類(這就是屏幕截圖中的所有內容)。 排名前的字母都是Domain,Kingdom,Phylum,Class,Order,Family,Genus和Species(分類法順序)的縮寫。 每行末尾的數字代表樣本中相應物種(或領域,門類等,如果未識別出)的比例。 該程序無法識別許多物種,因為尚未發現這些物種。
Visual & Categorical Analysis in R
R中的視覺和分類分析
After finally cleaning and classifying the raw data, we will move to R to visualize and categorize the data. Mystery DNA will be added to the dataset to simplify the purpose of using R, and our new goal will be to identify this data through visualization.
在最終清理和分類原始數據之后,我們將移至R以對數據進行可視化和分類。 神秘DNA將添加到數據集中,以簡化使用R的目的,而我們的新目標將是通過可視化識別此數據。
The first step in R Studio is to call the packages needed to visualize the data — ggplot2 is a package used to create all sorts of graphs and plots. The data is then read in using the read.csv function. Next, the geom_bar function of ggplot2 is used to create a barplot of the phylum classifications of the various samples:
R Studio的第一步是調用可視化數據所需的軟件包-ggplot2是用于創建各種圖形和繪圖的軟件包。 然后使用read.csv函數讀取數據。 接下來,使用ggplot2的geom_bar函數創建各種樣本的門類分類的barplot:
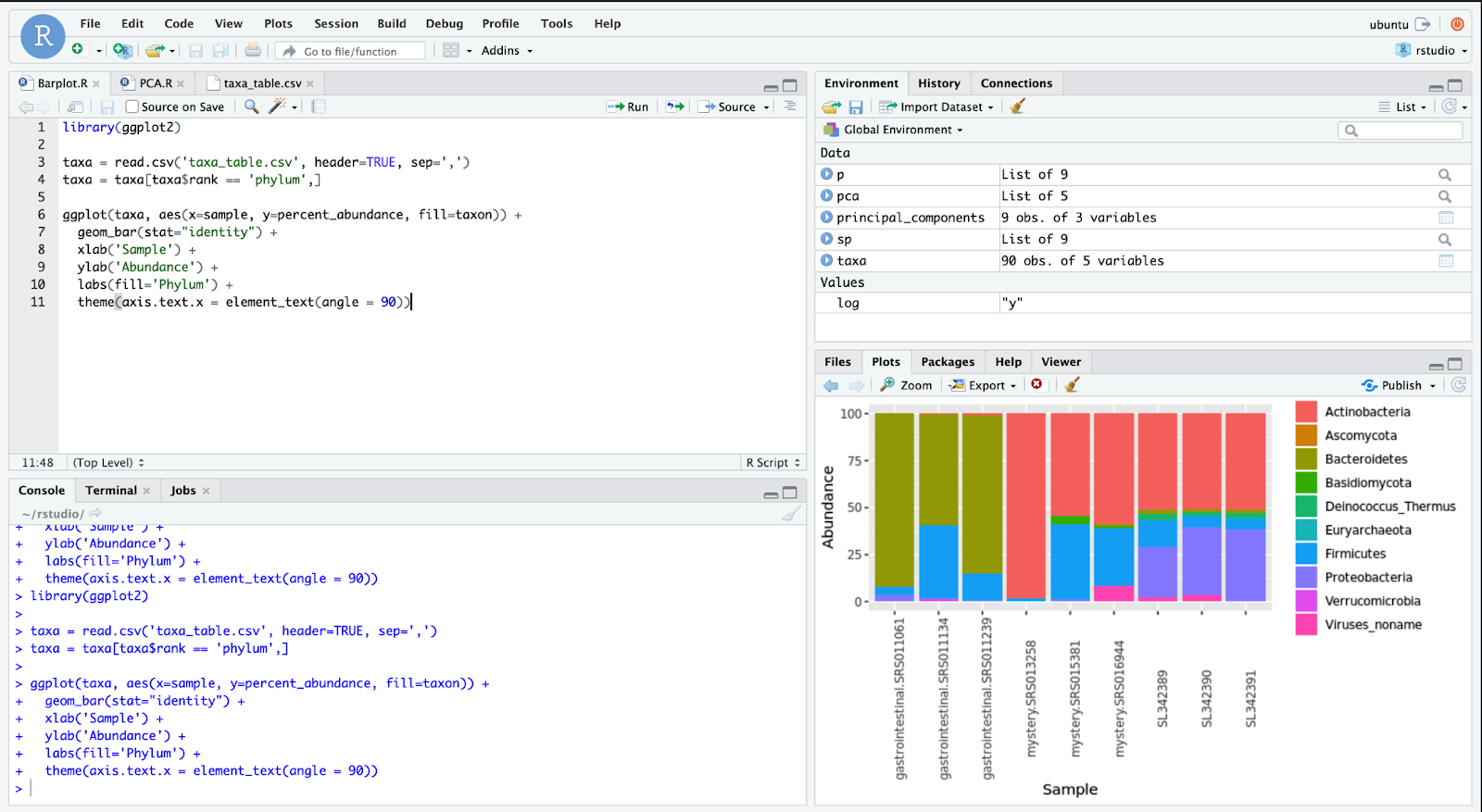
As shown in the bar graph from Figure 7, there are three samples from each environment — the first three are gastrointestinal data, then there are three samples from a mystery environment, and the final three come from shared transportation spaces. Already, some distinctions are apparent between the three groups. Gastrointestinal samples contain far more Bacteroidetes than the two other environments. The first mystery sample almost entirely consists of Actinobacteria. Despite these initial observations, it is difficult to come to a conclusion on the mystery samples since they are only grouped broadly by the microbes’ phyla, as seen in line 4 of the code. But before we make a bar graph of more specific taxonomic groups, it is necessary to analyze the variance among the samples. Principal Component Analysis (PCA), which decreases the number of variables or dimensions in the dataset, makes it easier to see the variance and patterns among groups in the dataset. As seen in Figure 8, some lines of code are added to create this PCA plot, and the points are colored according to their environment.
如圖7的條形圖所示,每個環境有三個樣本-前三個樣本是胃腸道數據,然后三個樣本來自一個神秘環境,最后三個樣本來自共享運輸空間。 這三組之間已經有一些明顯的區別。 胃腸道樣品中的擬桿菌含量遠高于其他兩種環境。 第一個神秘樣本幾乎完全由放線菌組成。 盡管有這些最初的觀察結果,但很難對神秘樣本做出結論,因為如代碼第4行所示,這些樣本僅按微生物的門類進行了大致分組。 但是在制作更具體的分類組的條形圖之前,有必要分析樣本之間的差異。 主成分分析(PCA)減少了數據集中變量或維的數量,使查看數據集中各組之間的差異和模式變得更加容易。 如圖8所示,添加了一些代碼行以創建此PCA圖,并且根據其環境為這些點著色。
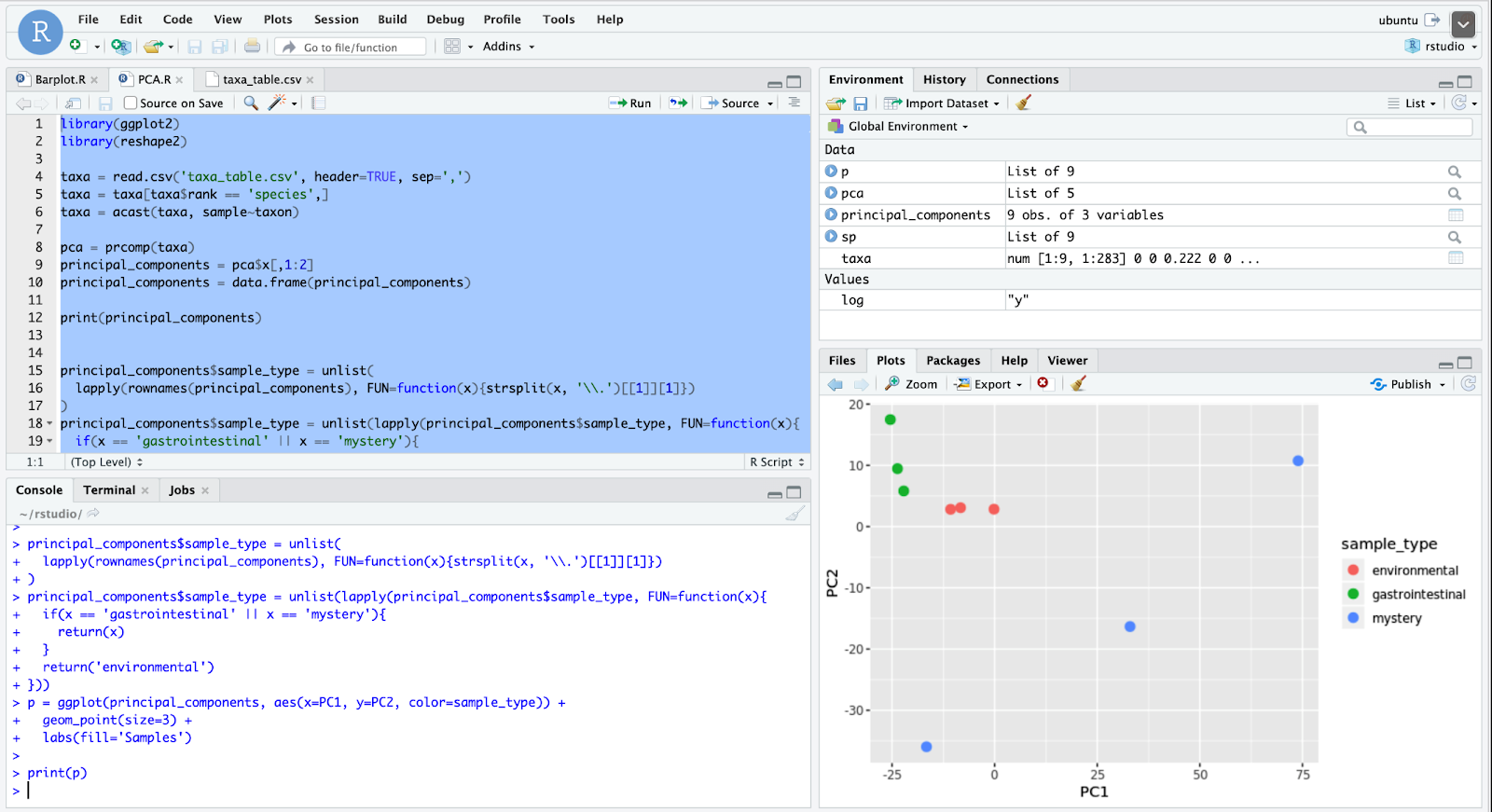
A few conclusions can be drawn from the plot in Figure 8. One is that both the environmental (mass transit) and gut samples lack variance since points are clustered tightly within each group. Meanwhile, mystery samples are not nearly as correlated since their points are spread apart, which tells us that those samples come from an environment where the types of microbes vary. From the initial conclusions, one might be quick to suggest that the environment is one of soil, as this environment is known to contain large amounts of actinobacteria and is also very variable. However, this conclusion is likely flawed since a more specific grouping of the first bar graph is needed to compare to other studies. When ranked by genus, the taxonomic abundances of the mystery samples look far more similar to the one on the right, which comes from a research paper on skin pore taxonomic abundance, not soil. Even though Figure 9 comes from a study on the impact of pomegranate juice on human skin microbiota, we can compare its control groups with our data as well.
從圖8中的曲線可以得出一些結論。一個是環境(大眾運輸)樣本和腸道樣本都缺乏方差,因為每個組中的點緊密聚集。 同時,由于神秘樣本的點分散開,因此它們之間的相關度也不高,這告訴我們這些樣本來自微生物種類不同的環境。 從最初的結論來看,可能會很快暗示環境是土壤的一種,因為已知該環境包含大量放線菌,而且變化很大。 但是,該結論可能有缺陷,因為需要將第一條形圖的更具體分組與其他研究進行比較。 當按屬進行排序時,神秘樣本的生物分類豐度看起來與右側樣本更加相似,后者來自有關皮膚 毛Kong生物分類豐度而不是土壤的研究論文。 即使圖9來自石榴汁對人類皮膚微生物群影響的研究,我們也可以將其對照組與我們的數據進行比較。
As it turns out, Propionibacterium and Staphylococcus were the most common microbes in the mystery samples. Still, we would lack this specificity without narrowing down the taxonomic grouping from phylum to genus. Indeed, human skin pores were the correct environment from which the mystery samples were taken.
事實證明,丙酸桿菌和葡萄球菌是神秘樣品中最常見的微生物。 但是,如果不縮小分類范圍,從門類到屬類,我們將缺乏這種特異性。 確實,人的皮膚毛Kong是從中取樣神秘樣品的正確環境。
Conclusions & Significance
結論與意義
Although the main visualization and analysis stages of this project were simplified compared to the actual processes of MetaSUB, projects like these bring light to the significance of their research. With so many unknown species lurking in mass transit spaces around the world, studying the microbiome can spur rapid innovation in the field. The possibilities of the research applications are endless — the data can play a crucial role in urban planning, city design, public health, and the discovery of new species. As the MetaSUB website says, the “data will… [enable] an era of more quantified, responsive, and smarter cities.” Especially with this past year’s COVID19 pandemic, researching and analyzing the microbial environments of spaces that billions of people share is all the more important, if not necessary.
盡管與MetaSUB的實際過程相比,該項目的主要可視化和分析階段得到了簡化,但類似的項目使他們的研究意義更為明顯。 由于世界各地的大眾運輸空間中潛伏著如此眾多的未知物種,因此研究微生物組可以促進該領域的快速創新。 研究應用的可能性是無限的-數據可以在城市規劃,城市設計,公共衛生和新物種發現中發揮關鍵作用。 正如MetaSUB網站所說,“數據將……(使)一個更量化,響應更快和更智能的城市時代”。 尤其是在去年的COVID19大流行中,研究和分析數十億人共享的空間的微生物環境,即使不是必須的話,也變得尤為重要。
Credits
學分
MILRD for providing the data, steps for the process, and invaluable mentorship.
MILRD用于提供數據,流程步驟和寶貴的指導。
Sources Used
資料來源
http://metasub.org/
http://metasub.org/
https://www.researchgate.net/figure/Relative-abundance-of-skin-microbiota-before-and-after-pomegranate-and-placebo_fig1_336383548
https://www.researchgate.net/figure/Relative-abundance-of-skin-microbiota-before-and-after-pomegranate-and-placebo_fig1_336383548
翻譯自: https://medium.com/swlh/microbial-surveillance-how-it-works-why-its-important-880f67aaa8b0
微生物 研究
本文來自互聯網用戶投稿,該文觀點僅代表作者本人,不代表本站立場。本站僅提供信息存儲空間服務,不擁有所有權,不承擔相關法律責任。 如若轉載,請注明出處:http://www.pswp.cn/news/389314.shtml 繁體地址,請注明出處:http://hk.pswp.cn/news/389314.shtml 英文地址,請注明出處:http://en.pswp.cn/news/389314.shtml
如若內容造成侵權/違法違規/事實不符,請聯系多彩編程網進行投訴反饋email:809451989@qq.com,一經查實,立即刪除!相關文章

Linux shell 腳本SDK 打包實踐, 收集assets和apk, 上傳FTP

opencv:卷積涉及的基礎概念,Sobel邊緣檢測代碼實現及卷積填充模式

怎么查這個文件在linux下的哪個目錄

無法從套接字中獲取更多數據_數據科學中應引起更多關注的一個組成部分

web數據交互_通過體育運動使用定制的交互式Web應用程序數據科學探索任何數據...

C# .net 對圖片操作

數據類型之Integer與int
思想及實現)
PCA(主成分分析)思想及實現

【安富萊二代示波器教程】第8章 示波器設計—測量功能

深度學習數據更換背景_開始學習數據科學的最佳方法是了解其背景

熊貓數據集_用熊貓掌握數據聚合

IOS CALayer的屬性和使用

GridView詳解

訪問模型參數,初始化模型參數,共享模型參數方法

QZEZ第一屆“飯吉圓”杯程序設計競賽

談談數據分析 caoz_讓我們談談開放數據…

數據創造價值_展示數據并創造價值

Java入門系列-22-IO流

