鏈特異性文庫是什么?為什么它在轉錄組測序中越來越重要?
在現代分子生物學研究中,RNA測序(RNA-seq) 是一種廣泛應用的技術,用于分析基因在不同條件下的表達情況。而在RNA-seq的眾多技術細節中,有一個“隱秘但關鍵”的環節——鏈特異性文庫構建(Strand-specific library preparation)。這項技術雖然聽起來有些專業,但它對結果的準確性有著重要影響。本文將通俗地介紹鏈特異性文庫的原理、作用、常見方法及數據分析注意事項。
1. 什么是“鏈特異性”?
DNA是一種雙鏈螺旋結構,由一條正義鏈(+鏈)和一條反義鏈(–鏈)構成。轉錄過程中,通常是由DNA的反義鏈(–鏈)作為模板合成mRNA,從而使mRNA序列與正義鏈一致(除了堿基T被替換為U)。
而在傳統的RNA-seq文庫構建中,RNA被打斷后逆轉錄成cDNA,再建庫測序,這個過程不會記錄RNA是來源于哪一條DNA鏈的信息。我們只知道這段RNA存在,但不知道其是源于正鏈還是反鏈。
鏈特異性文庫構建的目標是,在建庫過程中通過特定方法保留RNA原始的轉錄方向性信息,從而區分每一條RNA是由正鏈還是反鏈轉錄來的。
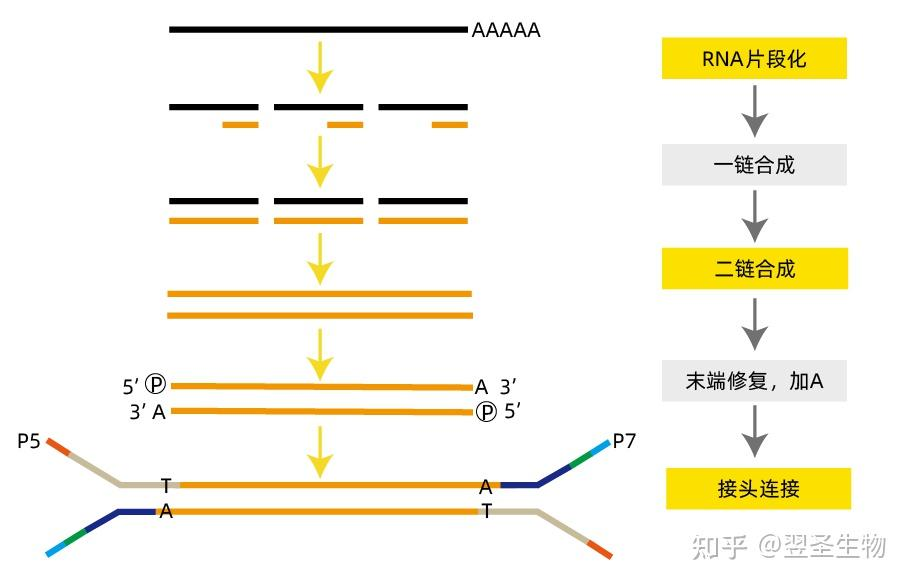
2. 為什么需要鏈特異性文庫?
鏈方向的保留,在多種分析中具有不可替代的重要性:
區分重疊基因
部分基因在基因組中是反向重疊的,即它們位于同一個基因組區域的兩條鏈上。如果沒有鏈信息,無法準確判斷這段表達信號來自哪個基因。
注釋非編碼RNA
例如lncRNA、反義轉錄本等非編碼RNA,常與編碼基因反向重疊。鏈信息是這些轉錄本精確注釋的關鍵。
提高定量精度
當多個基因之間位置相近或有部分重疊時,鏈特異性測序可顯著減少表達混淆,提高定量和差異分析的準確性。
3. 鏈特異性文庫的實現原理
主流鏈特異性文庫構建方法主要分為以下幾類,它們的共同目標是在建庫過程中保留或標記RNA的方向性信息。
方法一:dUTP法(Illumina常用方案)
dUTP法是目前最常用的鏈特異性建庫策略,原理如下:
- 合成第一鏈cDNA(使用mRNA為模板)
- 合成第二鏈時,用dUTP代替dTTP,使第二鏈中含有尿嘧啶(dU)
- 使用**UDG(Uracil-DNA Glycosylase)**選擇性降解含dU的第二鏈
- 僅保留第一鏈進行接頭連接與PCR建庫
此法操作簡單、成本低、兼容性好,是Illumina TruSeq等商業試劑盒的推薦方案。
注意事項:
- 測序得到的read方向與原始mRNA方向相反
- 常用參數方向性為
RF(Read1為反義)
方法二:接頭定向連接法(如 SMARTer、ScriptSeq)
通過在第一鏈cDNA末端引入方向性接頭或模板切換寡核苷酸(TSO),實現鏈信息的標記。例如:
- SMARTer法:只在第一鏈延伸出接頭,方向性由其控制。適用于低輸入甚至單細胞RNA。
- ScriptSeq法:通過特定引物和接頭組合區分方向,較早用于鏈特異性建庫。
方法三:標簽標記法(Ligation-based)
該法通過在cDNA兩端連接不同標簽序列來區分方向性,部分早期方案采用,但操作復雜,使用較少。
4. 如何判斷文庫是否為鏈特異性?
在測序實驗前或數據分析時,應確認建庫是否保留方向信息,可通過以下方法判斷:
- 查看實驗說明書或FastQC注釋,如“stranded = yes”
- 使用RSeQC工具(infer_experiment.py) 判斷read是否集中來源于特定鏈
- 檢查比對軟件中strand參數是否正確設置,避免方向誤判
5. 鏈特異性數據的分析注意事項
分析鏈特異性RNA-seq數據時,需明確方向性設定:
| 分析步驟 | 重點參數 | 示例說明 |
|---|---|---|
| 比對軟件 | 設置strand參數 | HISAT2示例:--rna-strandness RF |
| featureCounts計數工具 | 設定鏈信息 | -s 1為正鏈,-s 2為反鏈(dUTP法用-s 2) |
| HTSeq-count工具 | 設置為reverse方向 | -s reverse |
| 定量分析 | 匹配注釋方向 | lncRNA尤其敏感,方向錯會導致顯著誤判 |
6. 建庫方案選擇建議與參數配置
建庫方法建議
| 研究目標 | 建議建庫方案 | 說明 |
|---|---|---|
| mRNA表達分析 | dUTP法(TruSeq) | 成熟穩定、性價比高 |
| 非編碼RNA分析(lncRNA等) | dUTP法或SMARTer法 | 保留方向,適合復雜轉錄本識別 |
| 單細胞或低起始量樣本 | SMARTer、NEBNext Ultra II | 高靈敏度,適合微量RNA |
| 全轉錄組/非polyA分析 | rRNA去除 + dUTP法 | 可識別非polyA RNA轉錄本 |
實驗參數配置參考(以dUTP法為例)
| 步驟 | 參數或建議 |
|---|---|
| RNA輸入量 | 100 ng – 1 μg,依樣品而定 |
| 打斷條件 | 94°C,4–8分鐘,目標片段200–400 bp |
| 第一鏈合成 | 使用SuperScript II或III等高效酶 |
| 第二鏈合成 | 用dUTP替代dTTP |
| 降解第二鏈 | 使用USER酶去除含dU鏈 |
| PCR擴增 | 控制在10–15個cycle內 |
| 文庫質控 | Bioanalyzer檢測片段分布峰值約300 bp |
7. 數據分析參數設置示例
HISAT2 比對示例
hisat2 -x genome_index -1 R1.fastq -2 R2.fastq --rna-strandness RF
其中 RF 表示鏈特異性雙端測序,第一條read與mRNA方向相反。
STAR 比對配置
--outSAMstrandField intronMotif
--outSAMtype BAM SortedByCoordinate
--outFilterMultimapNmax 1
--twopassMode Basic
STAR支持鏈方向性,但后續需在featureCounts中設定方向。
featureCounts 示例
featureCounts -s 2 -p -T 8 -a annotation.gtf -o counts.txt aligned.bam
-s 2 表示反向鏈特異性,適用于dUTP建庫。
HTSeq-count 示例
htseq-count -f bam -s reverse -r pos aligned.bam annotation.gtf
8. 如何驗證鏈特異性是否有效
使用RSeQC工具包中的 infer_experiment.py 命令可以判斷測序數據是否保留鏈信息:
infer_experiment.py -i aligned.bam -r ref.bed
結果會輸出reads在不同鏈的分布比例。若某一類鏈向占比超過95%,說明鏈特異性建庫成功:
Fraction of reads explained by "1++,1--,2+-,2-+": 0.958
Fraction of reads explained by "1+-,1-+,2++,2--": 0.042
9. 常見方法與參數匯總
| 方法類型 | 建庫原理 | 分析參數方向性 | 適用場景 |
|---|---|---|---|
| dUTP法 | 第二鏈含dUTP并降解 | RF 或 -s 2 | 主流方案,Illumina推薦 |
| SMARTer法 | 模板切換接頭控制方向性 | 需自定義 | 低起始量或單細胞樣本 |
| Ligation-based法 | 接頭序列標記方向性 | 需自定義 | 特殊需求項目,較復雜較少使用 |
小結
鏈特異性文庫技術為RNA-seq分析帶來了更高的準確性,特別適用于區分反向重疊基因、識別非編碼RNA及提高定量精度。盡管建庫成本略有增加、分析參數需設定更精確,但其帶來的數據質量提升遠大于投入。如果你正計劃開展轉錄組研究,鏈特異性文庫無疑是值得優先選擇的建庫方式之一。
如需配套文庫構建圖示、參數設定流程圖、分析代碼封裝,歡迎留言交流。


系統及知識準備)
內存和內存地址、數組的查找算法和排序算法;)
場:河南農業大學(補題))

以pinia為中心的開發模板(監聽watch))








 :使用 Milvus 實現高效圖片查重功能)
的自述)


