***********************該筆記為初學者筆記,僅供個人參考謹慎搬運代碼******************************
samtools 排序壓縮和?featureCounts 生成基因計數表
SAM文件和BAM文件
1.SAM格式:是一種通用的比對格式,用來存儲reads到參考序列的比對信息,以TAB為分割符的文本格式。主要應用于測序序列mapping到基因組上的結果表示,當然也可以表示任意的多重比對結果。SAM分為兩部分,注釋信息和比對結果部分。
2.BAM格式:SAM的二進制文件,bam文件優點:bam文件為二進制文件,占用的磁盤空間比sam文本文件小;利用bam二進制文件的運算速度快。
#排序壓縮步驟
conda info --envs
conda activate py2env
#下載subread
conda install -c bioconda subread
#測試是否成功
featureCounts -h#成功
cd /home/yinwen/biosoft/hisat2
#壓縮:把 sam文件 轉為 bam文件
samtools view -S genome.sam -b > genome.bam
#排序:samtools sort
samtools sort -n -@ 5 genome.bam -o genome
#計數統計:
featureCounts -T 5 -t exon -g Parent -a genome.gff -o genome.counts -p genome
#samtools詳情:http://www.360doc.com/content/23/0927/14/1098188476_1098188476.shtml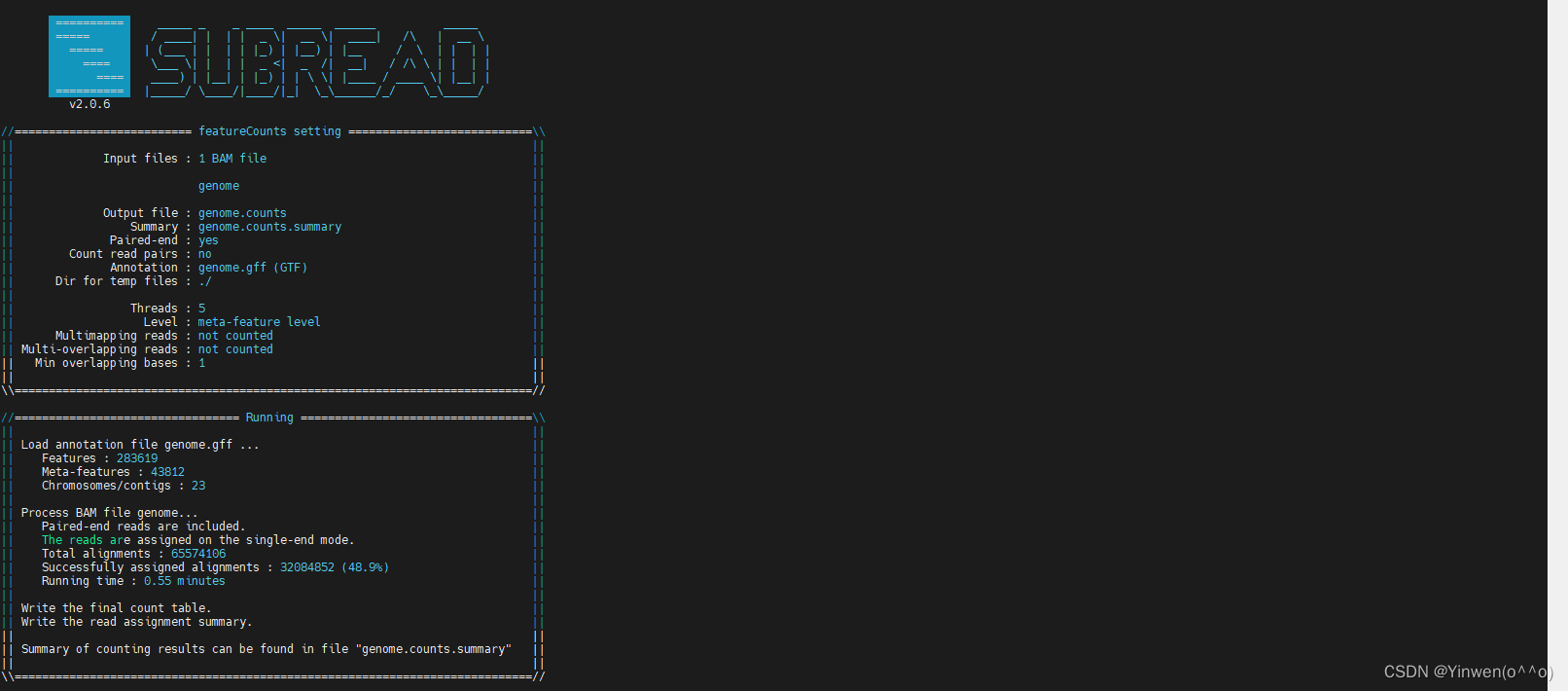
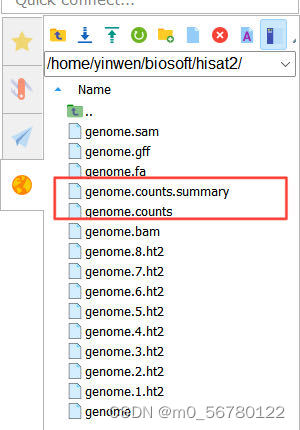
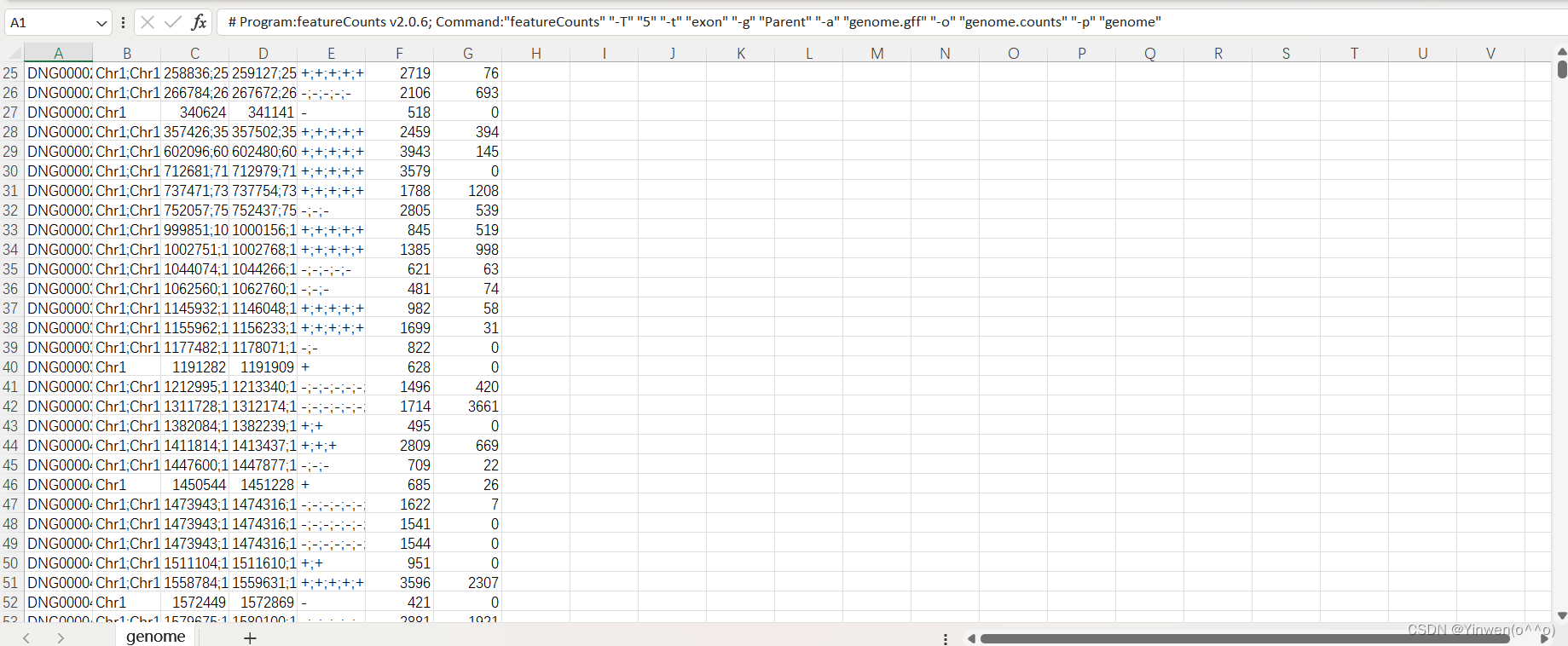
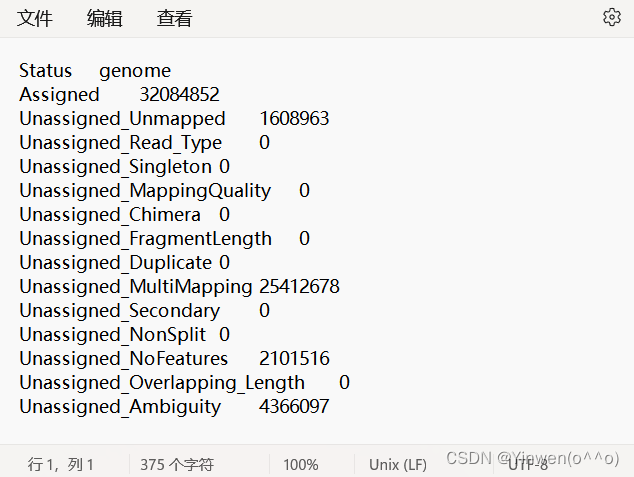
genome.gff 就是最初下載的注釋文件,如果要統計多個文件的話,在-p 后面跟上就可以,會生成 genome.counts、genome.counts.summary 兩個文件,我們的DG文件也是成功得到如下:
? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ??
求解23個基準函數(提供MATLAB代碼))
![BUUCTF---[極客大挑戰 2019]Upload1](http://pic.xiahunao.cn/BUUCTF---[極客大挑戰 2019]Upload1)
)








)

)





