本系列持續更新Seurat單細胞分析教程,歡迎關注!
非線形降維
Seurat 提供了幾種非線性降維技術,例如 tSNE 和 UMAP,來可視化和探索這些數據集。這些算法的目標是學習數據集中的底層結構,以便將相似的細胞放在低維空間中。因此,在上面確定的基于圖的簇內分組在一起的細胞應該在這些降維圖上共同定位。
雖然和其他人經常發現 tSNE 和 UMAP 等 2D 可視化技術是探索數據集的有價值的工具,但所有可視化技術都有局限性,并且不能完全代表基礎數據的復雜性。特別是,這些方法旨在保留數據集中的局部距離(即確保具有非常相似的基因表達譜的細胞共定位),但通常不會保留更多的全局關系。我們鼓勵用戶利用 UMAP 等技術進行可視化,但避免僅根據可視化技術得出生物學結論。
pbmc?<-?RunUMAP(pbmc,?dims?=?1:10)
#?note?that?you?can?set?`label?=?TRUE`?or?use?the?LabelClusters?function?to?help?label
#?individual?clusters
DimPlot(pbmc,?reduction?=?"umap")
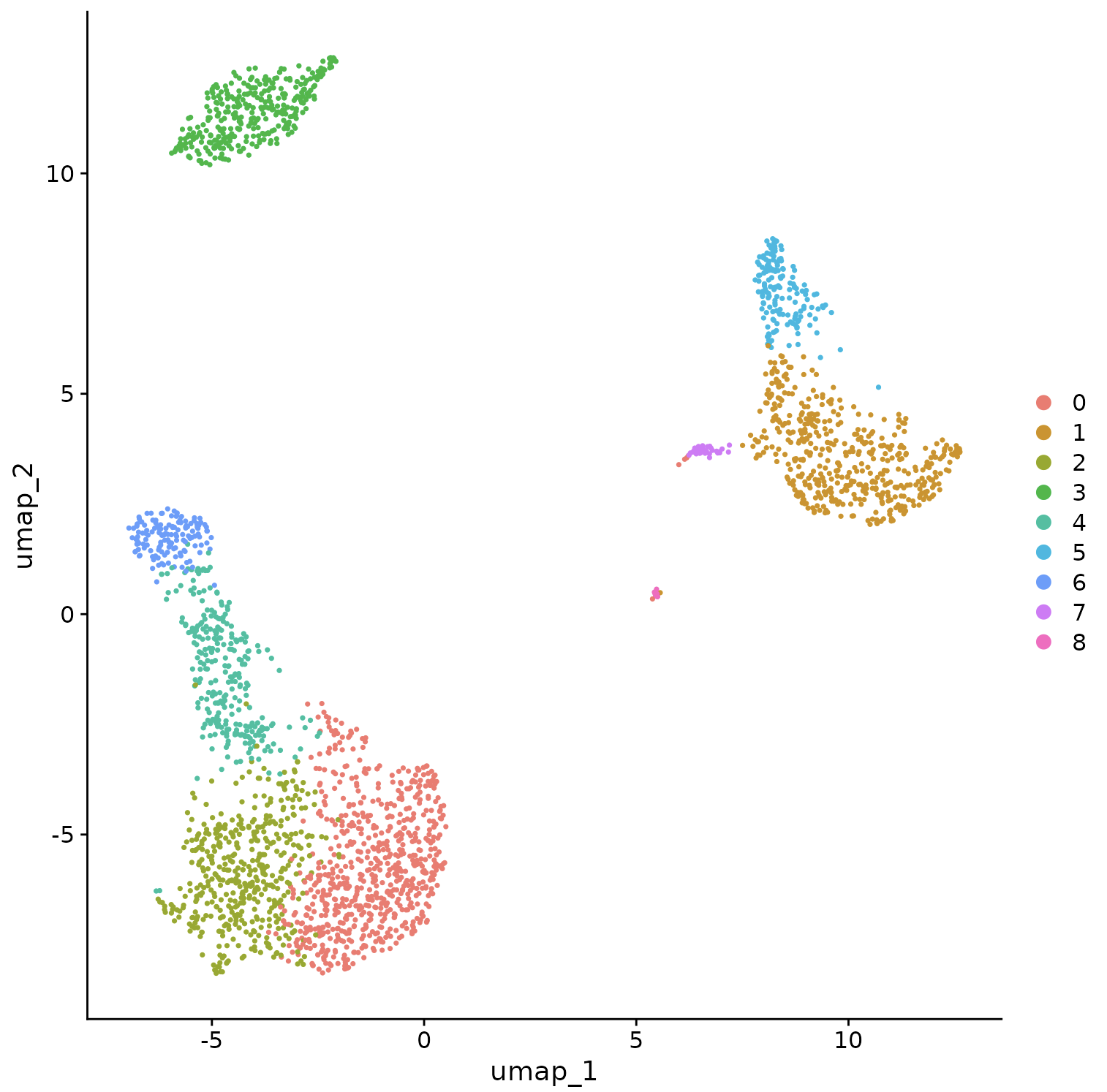
您可以在此時保存對象,以便可以輕松地重新加載它,而無需重新運行上面執行的計算密集型步驟,或者輕松地與協作者共享。
尋找差異表達特征(簇生物標志物)
Seurat 可以幫助您找到通過差異表達 (DE) 定義簇的標記。默認情況下,與所有其他細胞相比,它識別單個簇的陽性和陰性標記(在 ident.1 中指定)。 FindAllMarkers() 會針對所有集群自動執行此過程,但您也可以測試集群組之間的對比,或針對所有細胞進行測試。
在 Seurat v5 中,我們使用 presto 軟件包來顯著提高 DE 分析的速度,特別是對于大型數據集。對于不使用 presto 的用戶,您可以查看該函數的文檔(?FindMarkers)來探索 min.pct 和 logfc.threshold 參數,可以增加這些參數以提高 DE 測試的速度。
#?find?all?markers?of?cluster?2
cluster2.markers?<-?FindMarkers(pbmc,?ident.1?=?2)
head(cluster2.markers,?n?=?5)
##?????????????p_val?avg_log2FC?pct.1?pct.2????p_val_adj
##?IL32?2.593535e-91??1.3221171?0.949?0.466?3.556774e-87
##?LTB??7.994465e-87??1.3450377?0.981?0.644?1.096361e-82
##?CD3D?3.922451e-70??1.0562099?0.922?0.433?5.379250e-66
##?IL7R?1.130870e-66??1.4256944?0.748?0.327?1.550876e-62
##?LDHB?4.082189e-65??0.9765875?0.953?0.614?5.598314e-61
#?find?all?markers?distinguishing?cluster?5?from?clusters?0?and?3
cluster5.markers?<-?FindMarkers(pbmc,?ident.1?=?5,?ident.2?=?c(0,?3))
head(cluster5.markers,?n?=?5)
##???????????????????????p_val?avg_log2FC?pct.1?pct.2?????p_val_adj
##?FCGR3A????????2.150929e-209???6.832372?0.975?0.039?2.949784e-205
##?IFITM3????????6.103366e-199???6.181000?0.975?0.048?8.370156e-195
##?CFD???????????8.891428e-198???6.052575?0.938?0.037?1.219370e-193
##?CD68??????????2.374425e-194???5.493138?0.926?0.035?3.256286e-190
##?RP11-290F20.3?9.308287e-191???6.335402?0.840?0.016?1.276538e-186
#?find?markers?for?every?cluster?compared?to?all?remaining?cells,?report?only?the?positive
#?ones
pbmc.markers?<-?FindAllMarkers(pbmc,?only.pos?=?TRUE)
pbmc.markers?%>%
????group_by(cluster)?%>%
????dplyr::filter(avg_log2FC?>?1)
##?#?A?tibble:?7,046?×?7
##?#?Groups:???cluster?[9]
##????????p_val?avg_log2FC?pct.1?pct.2?p_val_adj?cluster?gene?????
##????????<dbl>??????<dbl>?<dbl>?<dbl>?????<dbl>?<fct>???<chr>????
##??1?1.74e-109???????1.19?0.897?0.593?2.39e-105?0???????LDHB?????
##??2?1.17e-?83???????2.37?0.435?0.108?1.60e-?79?0???????CCR7?????
##??3?8.94e-?79???????1.09?0.838?0.403?1.23e-?74?0???????CD3D?????
##??4?3.05e-?53???????1.02?0.722?0.399?4.19e-?49?0???????CD3E?????
##??5?3.28e-?49???????2.10?0.333?0.103?4.50e-?45?0???????LEF1?????
##??6?6.66e-?49???????1.25?0.623?0.358?9.13e-?45?0???????NOSIP????
##??7?9.31e-?44???????2.02?0.328?0.11??1.28e-?39?0???????PRKCQ-AS1
##??8?4.69e-?43???????1.53?0.435?0.184?6.43e-?39?0???????PIK3IP1??
##??9?1.47e-?39???????2.70?0.195?0.04??2.01e-?35?0???????FHIT?????
##?10?2.44e-?33???????1.94?0.262?0.087?3.34e-?29?0???????MAL??????
##?#???7,036?more?rows
Seurat 有幾種差異表達測試,可以使用 test.use 參數進行設置。例如,ROC 測試返回任何單個標記的“分類能力”(范圍從 0 到 1)。
cluster0.markers?<-?FindMarkers(pbmc,?ident.1?=?0,?logfc.threshold?=?0.25,?test.use?=?"roc",?only.pos?=?TRUE)
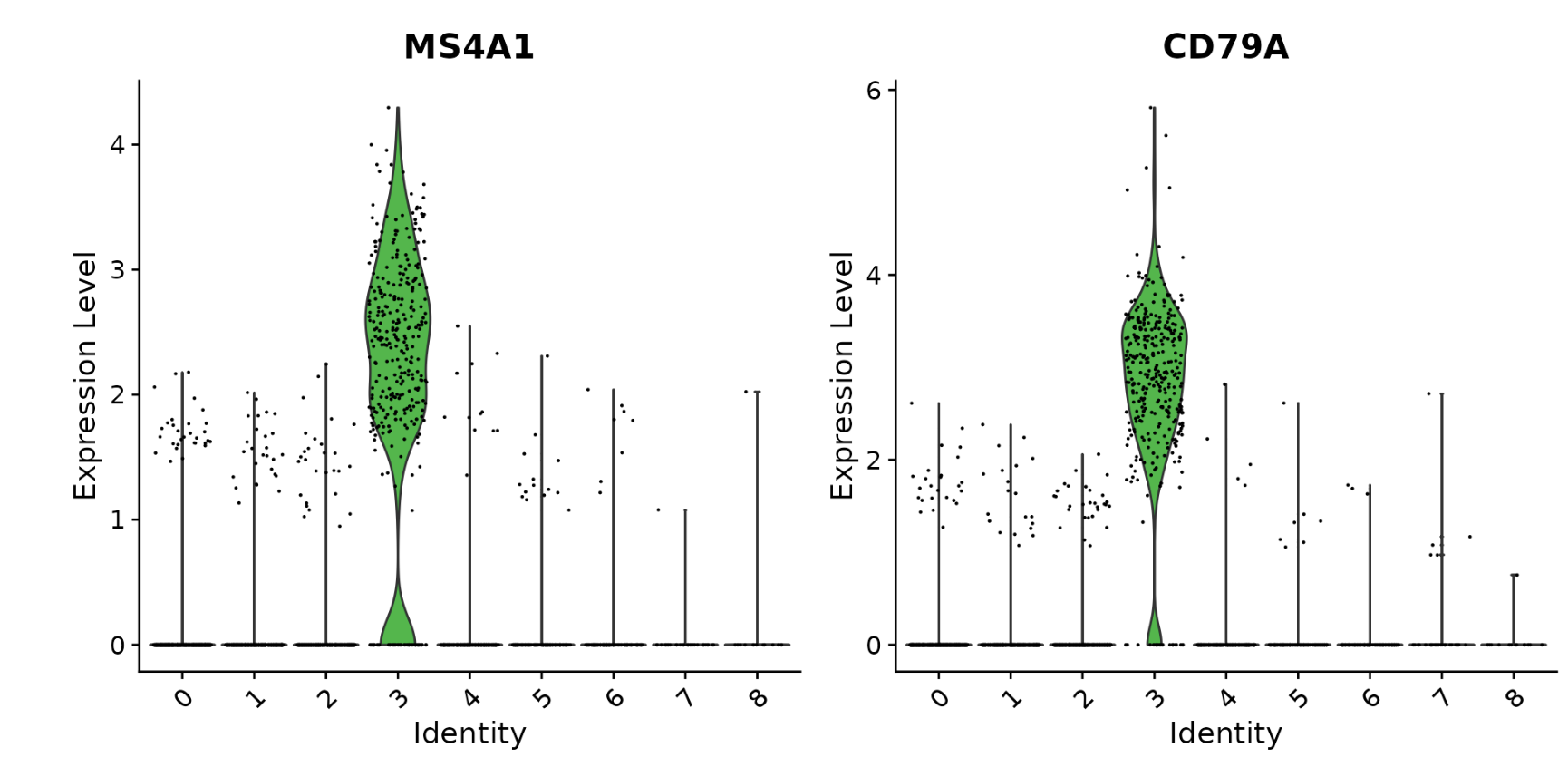
我們提供了幾種用于可視化標記表達的工具。 VlnPlot()(顯示跨簇的表達概率分布)和 FeaturePlot()(在 tSNE 或 PCA 圖上可視化特征表達)是我們最常用的可視化。我們還建議探索 RidgePlot()、CellScatter() 和 DotPlot() 作為查看數據集的附加方法。
VlnPlot(pbmc,?features?=?c("MS4A1",?"CD79A"))
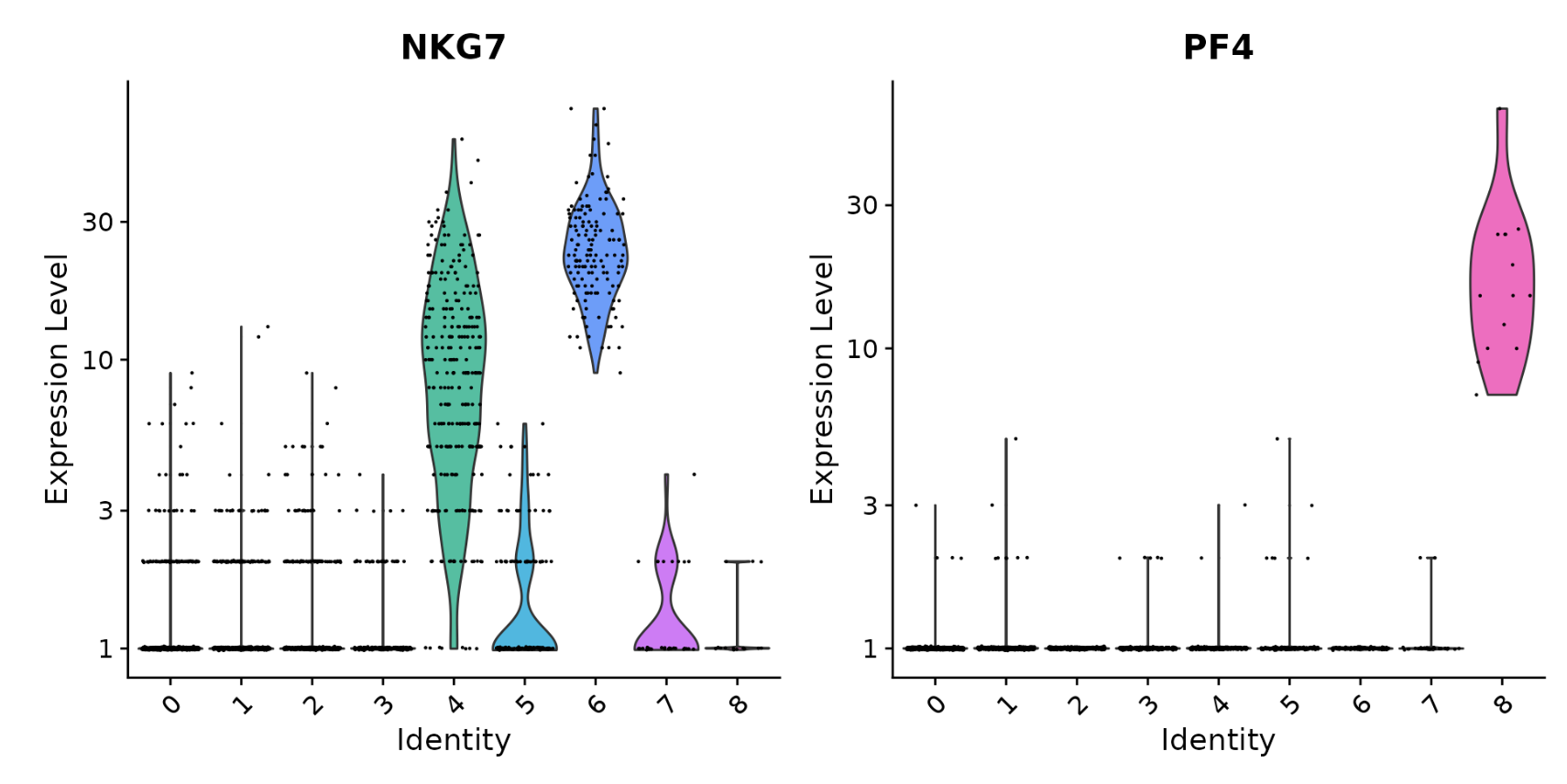
#?you?can?plot?raw?counts?as?well
VlnPlot(pbmc,?features?=?c("NKG7",?"PF4"),?slot?=?"counts",?log?=?TRUE)
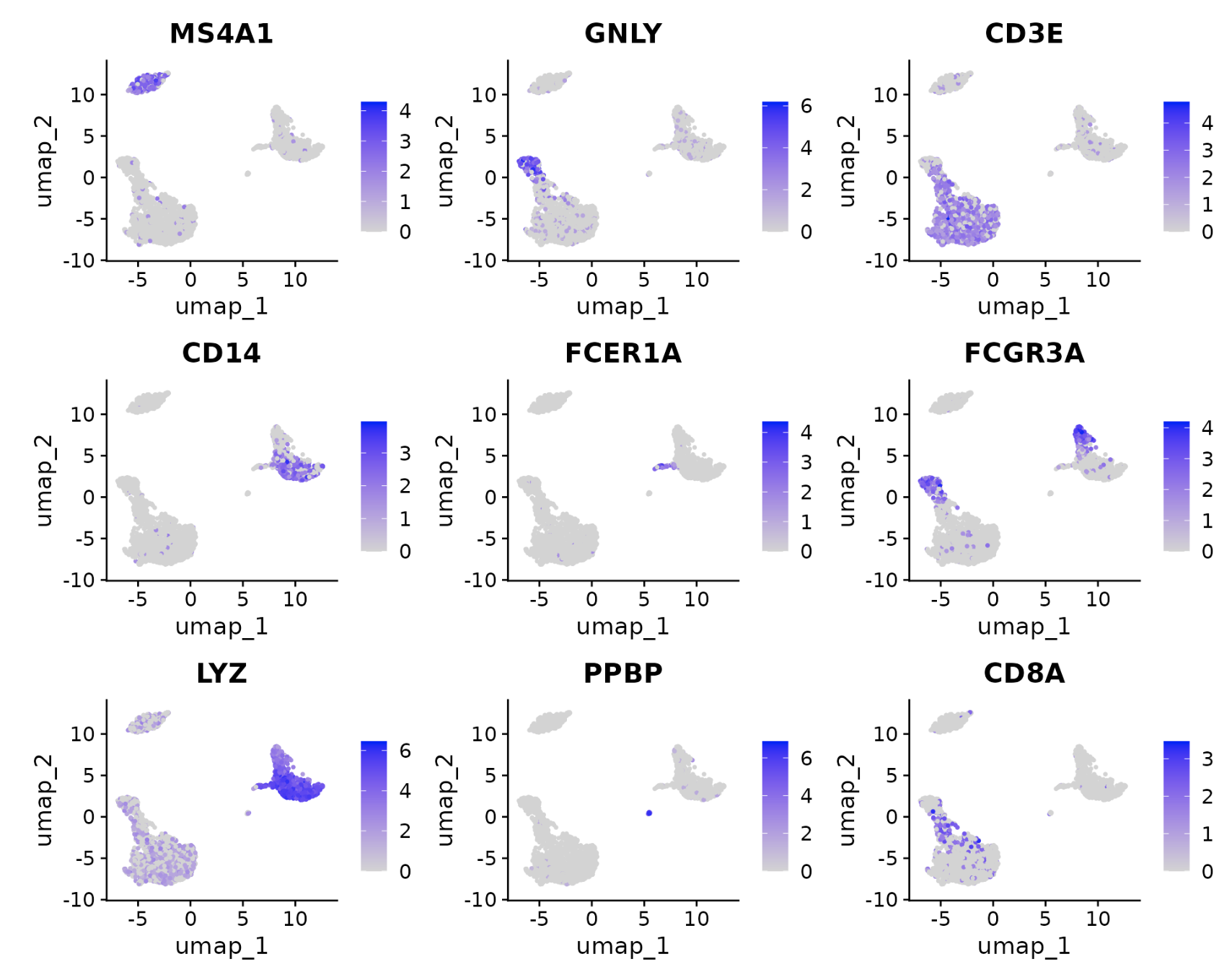
FeaturePlot(pbmc,?features?=?c("MS4A1",?"GNLY",?"CD3E",?"CD14",?"FCER1A",?"FCGR3A",?"LYZ",?"PPBP",
????"CD8A"))
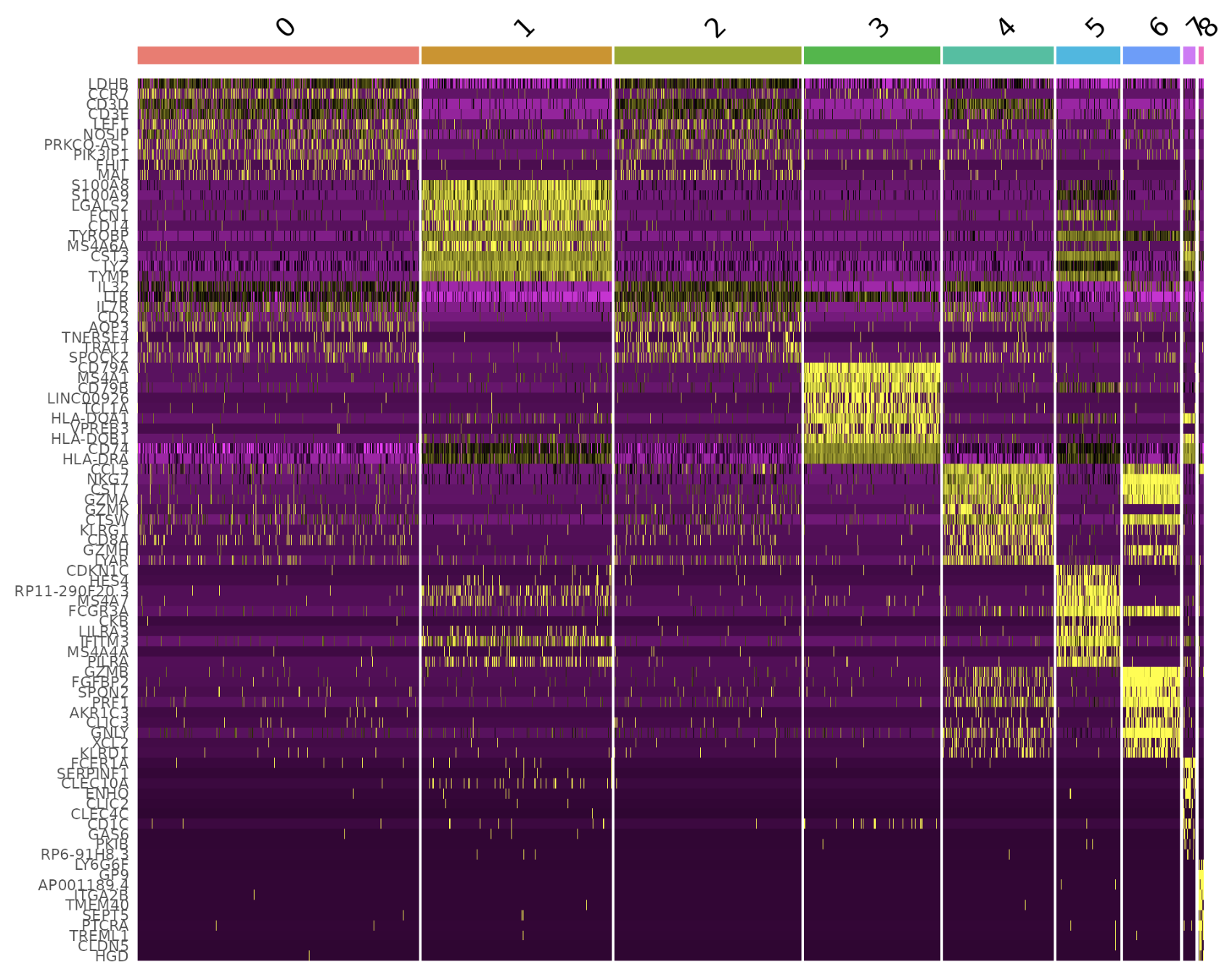
DoHeatmap() 為給定的細胞和特征生成表達式熱圖。在本例中,我們繪制每個簇的前 20 個標記(如果少于 20 個則為所有標記)。
pbmc.markers?%>%
????group_by(cluster)?%>%
????dplyr::filter(avg_log2FC?>?1)?%>%
????slice_head(n?=?10)?%>%
????ungroup()?->?top10
DoHeatmap(pbmc,?features?=?top10$gene)?+?NoLegend()
細胞類型分配
在此數據集的情況下,可以使用規范標記輕松地將無偏聚類與已知細胞類型進行匹配:
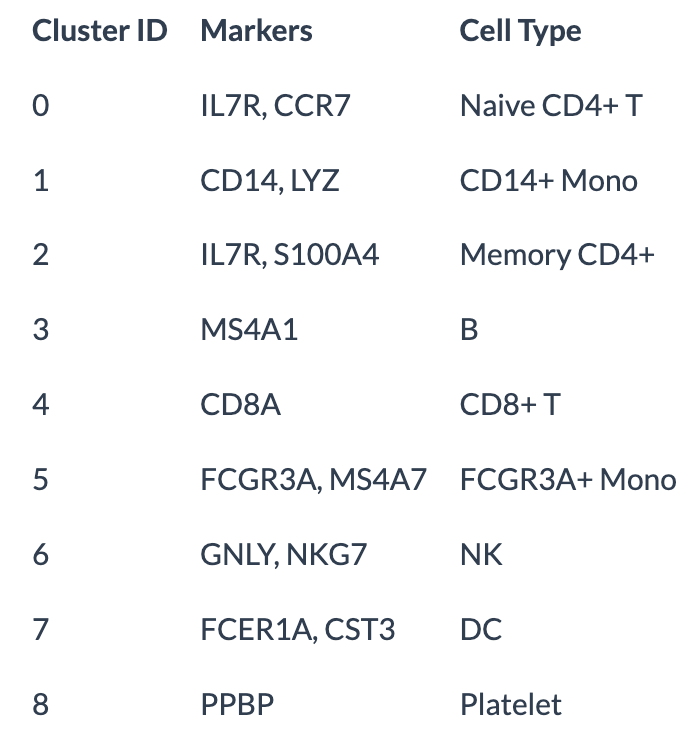
new.cluster.ids?<-?c("Naive?CD4?T",?"CD14+?Mono",?"Memory?CD4?T",?"B",?"CD8?T",?"FCGR3A+?Mono",
????"NK",?"DC",?"Platelet")
names(new.cluster.ids)?<-?levels(pbmc)
pbmc?<-?RenameIdents(pbmc,?new.cluster.ids)
DimPlot(pbmc,?reduction?=?"umap",?label?=?TRUE,?pt.size?=?0.5)?+?NoLegend()
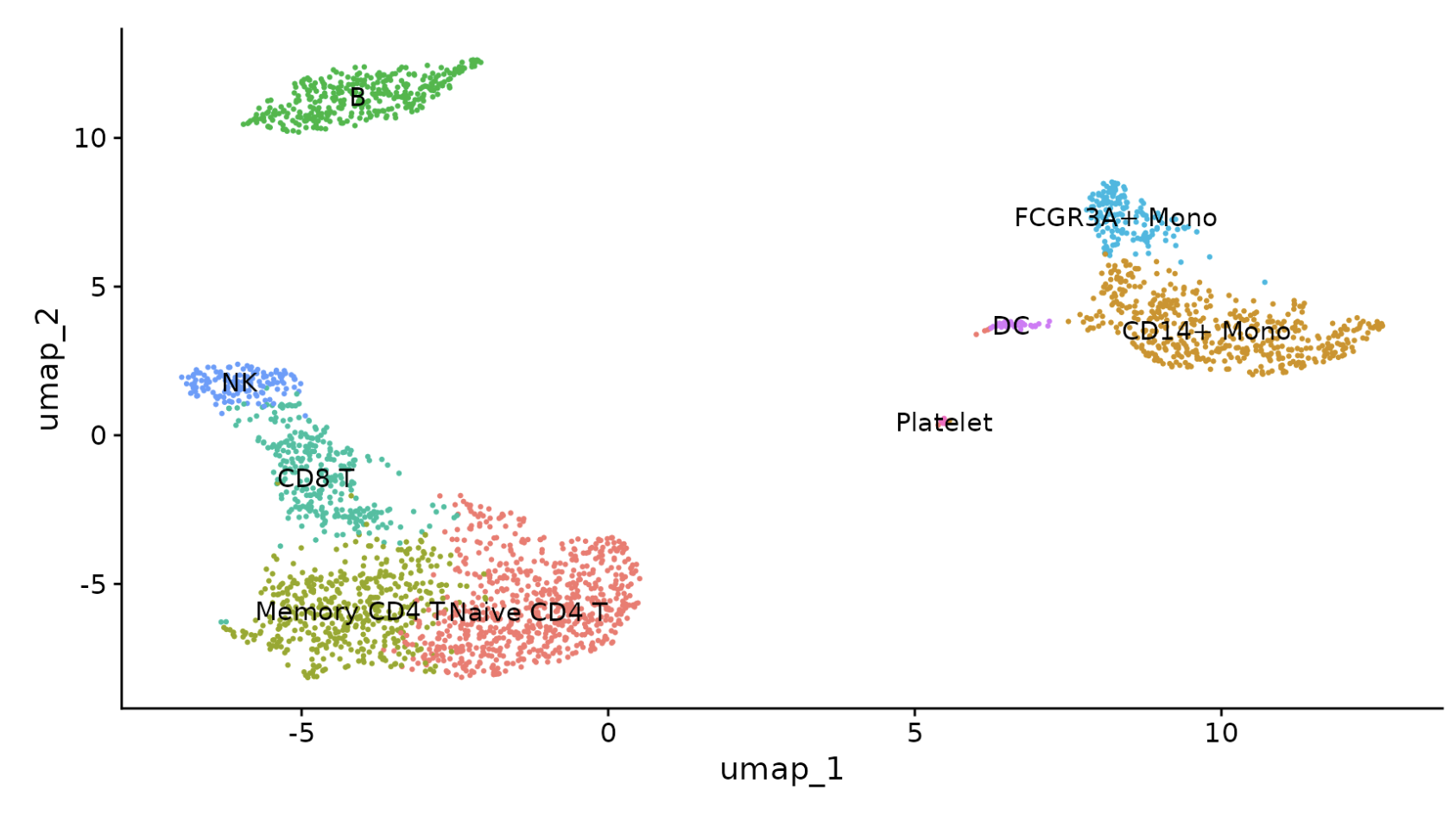
library(ggplot2)
plot?<-?DimPlot(pbmc,?reduction?=?"umap",?label?=?TRUE,?label.size?=?4.5)?+?xlab("UMAP?1")?+?ylab("UMAP?2")?+
????theme(axis.title?=?element_text(size?=?18),?legend.text?=?element_text(size?=?18))?+?guides(colour?=?guide_legend(override.aes?=?list(size?=?10)))
ggsave(filename?=?"../output/images/pbmc3k_umap.jpg",?height?=?7,?width?=?12,?plot?=?plot,?quality?=?50)
saveRDS(pbmc,?file?=?"../output/pbmc3k_final.rds")
未完待續,持續更新,歡迎關注!
本文由 mdnice 多平臺發布

)

機器人柵格路徑規劃,輸出做短路徑圖和適應度曲線。)

)



)





)
)

)
