1. 引言:單細胞數據整合的背景與重要性
單細胞基因組學技術(如scRNA-seq、scATAC-seq等)近年來快速發展,能夠以單細胞分辨率揭示細胞異質性和分子機制。然而,不同實驗、樣本和數據模態(如RNA表達、DNA甲基化、蛋白質分布等)之間存在技術變異和生物學差異,這使得數據整合成為單細胞研究的關鍵挑戰。文章可能從以下幾個方面探討單細胞數據整合的計算原理和面臨的統計與技術難題:
- 研究目標:通過整合多源單細胞數據,消除技術噪聲,保留生物學信號,從而構建統一的細胞狀態圖譜,支持跨實驗、跨物種和跨模態的分析。
- 應用場景:包括疾病研究(如癌癥、免疫相關疾病)、發育生物學(如胚胎發育跨物種比較)以及個性化醫學中的患者分層。
2. 數據整合的主要類型
 ???????
???????
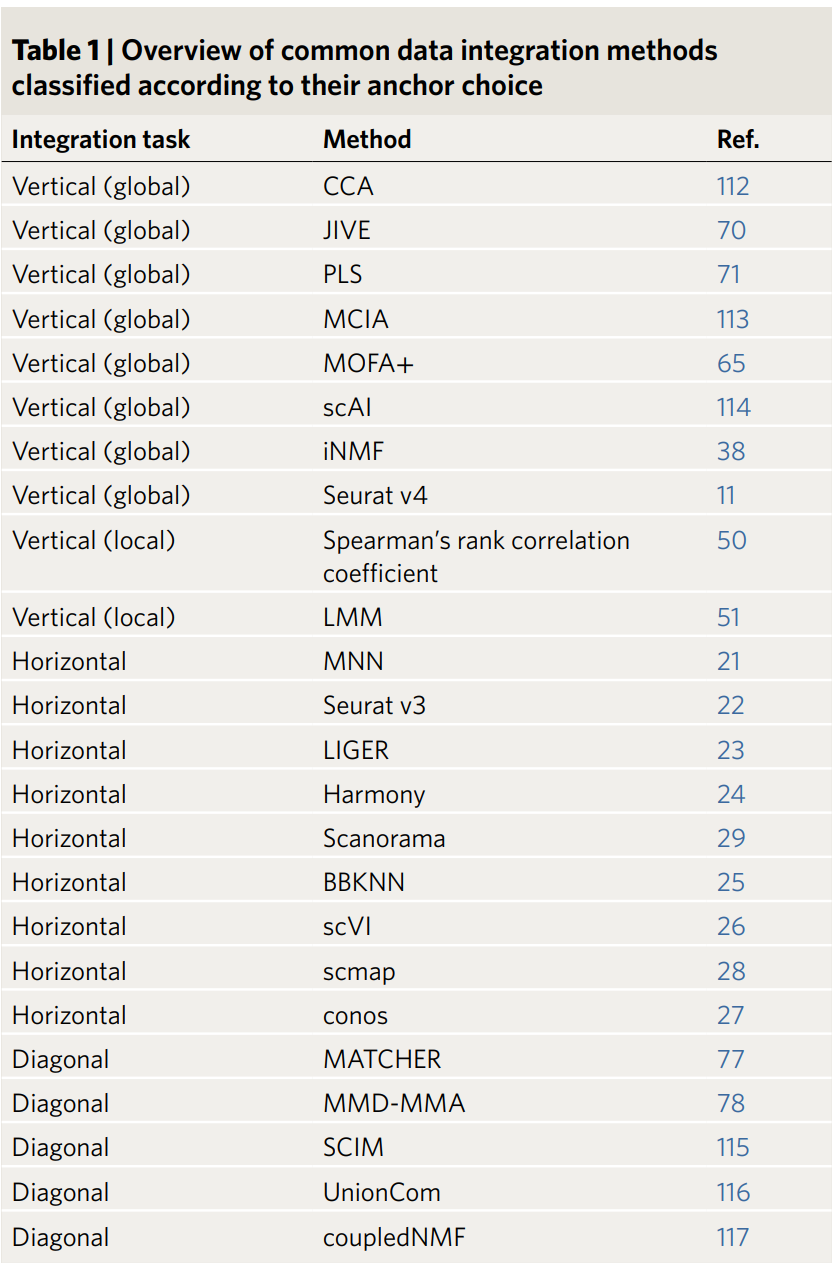
Table 1
根據文檔中的表格(Table 1)和描述,文章可能系統性地分類了單細胞數據整合的主要任務,基于“錨點選擇”(anchor choice)分為以下幾類:
垂直整合(Vertical Integration):
全局垂直整合(Vertical Global):整合同一實驗或數據集內不同模態的數據(如RNA表達和表觀遺傳數據),以揭示多組學層面上的細胞狀態。
???????????????全局垂直整合就像把所有照片疊在一起,找到每張照片中對應物體的共同特征(如形狀、位置),然后畫一張統一的“全景圖”,讓所有物體的位置和關系都能在同一張圖上清晰呈現。
????????工作原理
- ?輸入數據:通常需要配對的多組學數據(paired multi-omics data),即在同一細胞上測量多種模態的數據。例如,10X Multiome 技術可以同時生成一個細胞的 scRNA-seq(基因表達)和 scATAC-seq(染色質開放性)數據。這些配對數據作為“錨點”(anchors),幫助模型學習模態間的對應關系。
處理流程:
數據預處理:對不同模態數據進行標準化(例如,scRNA-seq 數據進行對數轉換,scATAC-seq 數據歸一化為基因活性評分),以統一統計特性。
降維或嵌入:通過算法將高維數據(例如,數千基因或數十萬峰值)映射到低維空間(如幾十維),使同一細胞在不同模態的表示盡可能接近。
優化目標:最小化模態間表示的距離,同時保留細胞類型或狀態的生物學差異。
常用方法:
典型相關分析(CCA):Seurat v3 使用 CCA 找到基因表達和染色質開放性之間的最大相關性,將數據投影到共享子空間。
非負矩陣分解(NMF):Liger 通過分解數據矩陣,提取共享因子(如細胞類型相關的基因模塊)。
多組學因子分析(MOFA+):通過潛在因子建模,捕捉多模態數據的共享變異。
深度學習方法:如變分自編碼器(VAE)或孿生網絡(Siamese Network)。例如,MinNet(Lu et al., 2023)使用孿生網絡,通過圖基對比損失(graph-based contrastive loss)優化嵌入空間。?????????????????????
局部垂直整合(Vertical Local):聚焦于特定細胞類型或子集的整合(整合多模態數據),可能用于局部特征的分析。
????????局部垂直整合就像只關注照片中的某一部分(如人群中的某幾個人),將這些人的彩色和黑白照片疊在一起,分析他們的具體特征(如面部表情、衣著),而忽略其他部分。目標是深入研究這部分人的細節,而不是整個場景。
????????工作原理
- 輸入數據:與全局整合類似,通常使用配對多組學數據,但只針對預先定義的細胞子集(如通過聚類或標記物基因識別的 T 細胞)。
- 處理流程:
- 子集選擇:通過聚類算法(如基于 scRNA-seq 的 Louvain 聚類)或生物標記物(如 T 細胞的 CD3 基因),篩選目標細胞子集。
- 局部嵌入:對子集數據應用降維或嵌入方法,生成局部的統一表示空間。
- 特征分析:在局部嵌入空間中,計算基因表達與調控元件之間的相關性,推斷局部調控網絡。
- 常用方法:
- 子集 CCA:在選定子集上應用 CCA,整合多模態數據。
- 局部 NMF:對子集數據進行矩陣分解,提取局部共享因子。
- 深度學習:如 MinNet 的子集應用,通過圖基對比損失優化局部嵌入空間。
全局垂直整合 VS 局部垂直整合
| 特性 | 全局垂直整合 | 局部垂直整合 |
|---|---|---|
| 目標 | 整合整個數據集,構建全面細胞狀態圖譜 | 聚焦特定細胞子集,分析局部生物學特征 |
| 數據范圍 | 所有細胞的多模態數據 | 選定子集的多模態數據 |
| 應用場景 | 細胞圖譜構建、疾病機制研究、發育軌跡分析 | 特定細胞類型分析、調控網絡推斷、疾病研究 |
| 計算復雜度 | 高,需處理大規模數據 | 較低,但可能需多次局部整合 |
| 主要挑戰 | 高維數據、模態異質性、過擬合、計算成本 | 子集選擇偏差、數據稀疏性、一致性問題 |
| 常用方法 | CCA、NMF、MOFA+、深度學習 | 子集 CCA、局部 NMF、深度學習 |
| 輸出 | 全局嵌入空間,適用于廣義分析 | 局部嵌入空間,聚焦特定生物學問題 |
水平整合(Horizontal Integration):主要解決批次效應(batch effect),即去除不同實驗或技術平臺之間的技術變異,以確保跨數據集的比較一致性。文章提到水平整合常被定義為“批次校正問題”(batch correction problem)。
????????目標是把來自不同來源(比如不同實驗室、不同患者、不同時間點)的同一種單細胞數據(通常是 scRNA-seq,也可能是 scATAC-seq 或 CITE-seq)整合到一個統一的表示空間(embedding space)。這個空間里,相同類型的細胞(比如 T 細胞)應該聚在一起,不管它們來自哪個實驗,而不同類型的細胞(比如 T 細胞和 B 細胞)應該分開。
即將不同來源的同一個類型的東西放在一起。注:處理同一種模態的數據。
工作原理
- 輸入數據:多組同模態數據(比如幾組 scRNA-seq 數據),每組來自不同樣本、實驗或條件。數據通常是基因表達矩陣(行是基因,列是細胞)。
- 預處理:
- 標準化:對每組數據進行歸一化(比如對數轉換、單位方差歸一化),統一數據分布。
- 特征選擇:挑出高變異基因(highly variable genes),減少噪音和計算量。
- 整合方法:
- 找到每組數據中的“錨點”(anchors),即可能對應相同細胞類型的細胞對。
- 將所有數據投影到一個共享的低維空間,確保錨點對齊。
- 優化目標:最小化不同數據集間的技術差異(批次效應),最大化生物學差異(比如細胞類型分離)。
- 輸出:一個統一的低維嵌入空間(比如用 PCA 或 UMAP 降維后的空間),所有細胞都在這個空間里表示為點,相同細胞類型聚在一起。
常用方法:
- Seurat v3:用**典型相關分析(CCA)或互近鄰(Mutual Nearest Neighbors, MNN)**找錨點,把不同數據集的細胞對齊。
- Harmony:用一種迭代聚類方法,逐步校正批次效應,生成統一的嵌入空間。
- Scanorama:基于 MNN 匹配細胞,校正批次差異。
- Liger:用非負矩陣分解(NMF)提取共享因子,整合不同數據集。
- scVI:用深度學習的變分自編碼器(VAE)建模數據分布,校正批次效應。
???????例子講解:
假設你在研究癌癥,想整合三組 scRNA-seq 數據:一組是健康人的 PBMC,一組是肺癌患者的腫瘤微環境細胞,一組是乳腺癌患者的腫瘤細胞。你想比較這些樣本里的免疫細胞狀態。
用 Seurat v3 做水平整合的步驟:
- 讀數據:把三組 scRNA-seq 數據讀進來,每個數據集是一個基因表達矩陣。
- 預處理:對每組數據做對數轉換,選出 2000 個高變異基因。
- 找錨點:用 CCA 找每組數據里可能對應的細胞(比如健康和癌癥樣本里的 T 細胞)。
- 整合:把三組數據投影到一個共享的 PCA 空間,校正批次效應。
- 可視化:用 UMAP 把嵌入空間畫出來,看看 T 細胞、B 細胞等是否聚成相同的簇。
- 分析:比較健康和癌癥樣本里 T 細胞的基因表達差異,找癌癥相關的免疫變化。
結果是:你得到一張 UMAP 圖,健康和癌癥樣本的 T 細胞聚在一起,B 細胞聚在一起,但癌癥樣本的 T 細胞可能表達一些特異基因(比如 PD-1,提示免疫抑制)。
應用場景:
- 疾病比較:整合健康和疾病樣本的數據,研究疾病引起的細胞變化。比如,整合 COVID-19 患者和健康人的 scRNA-seq 數據,找免疫細胞的差異。
- 跨物種研究:整合人和小鼠的 scRNA-seq 數據,找保守的細胞類型或基因模塊。
- 多實驗室協作:把不同實驗室用不同設備測的 scRNA-seq 數據整合,生成統一數據集。
- 時間序列分析:整合不同時間點的 scRNA-seq 數據,研究細胞發育或疾病進展的動態變化。
???????
對角整合(Diagonal Integration):可能涉及跨物種或跨時間點的整合(如文檔中提到的鼠和人胚胎發育的單細胞基因組學實驗)。對角整合的目標是整合多個樣本或條件下的多種模態數據(比如 scRNA-seq、scATAC-seq、CITE-seq),生成一個統一的低維嵌入空間(co-embedding space)。在這個空間里:
- 同一個細胞的多模態數據(比如基因表達和染色質開放性)應該盡可能接近(垂直整合的目標)。
- 不同樣本或條件的相同細胞類型(比如不同患者的 T 細胞)應該聚在一起(水平整合的目標)。
????????對角整合是同時整合不同樣本或條件的不同模態數據(比如多個患者的 scRNA-seq 和 scATAC-seq 數據),目標是消除批次效應(不同樣本的差異)和模態效應(不同數據類型的差異),生成一個統一的表示空間,讓相同細胞類型的多組學特征對齊。
工作原理
- 輸入數據:
- 多組數據,每組包含不同樣本或條件的多種模態數據。比如,三個患者的 scRNA-seq 和 scATAC-seq 數據,每個數據集是一個矩陣(行是基因/峰值,列是細胞)。
- 數據可能是配對的(paired,比如 Multiome 技術測的同一個細胞的兩種數據)或非配對的(unpaired,比如 scRNA-seq 和 scATAC-seq 來自不同細胞)。
- 預處理:
- 標準化:對每種模態數據進行歸一化(比如 scRNA-seq 對數轉換,scATAC-seq 轉為基因活性評分),統一統計特性。
- 特征選擇:選高變異基因(scRNA-seq)或高變異峰值(scATAC-seq),減少噪音和計算量。
- 批次標注:給每個樣本標記批次標簽(比如患者 1、患者 2),用于校正批次效應。
- 整合方法:
- 找錨點:在不同樣本和模態間找到對應的細胞對(錨點),比如通過互近鄰(MNN)或典型相關分析(CCA)匹配可能屬于同一細胞類型的細胞。
- 投影到共享空間:將所有數據(多樣本、多模態)映射到一個統一的低維空間,確保同一細胞的多種模態表示接近,同一細胞類型的不同樣本表示聚在一起。
- 校正效應:同時消除批次效應(樣本間差異)和模態效應(數據類型差異)。
- 優化目標:
- 模態對齊:最小化同一細胞在不同模態間的表示距離(比如用歐幾里得距離或 KL 散度)。
- 批次校正:最小化不同樣本間的技術差異,確保相同細胞類型對齊。
- 生物學信號保留:最大化細胞類型或狀態的分離(比如用分類損失確保 T 細胞和 B 細胞分開)。
- 輸出:一個統一的低維嵌入空間,所有細胞(無論來自哪個樣本或模態)都表示為點,同一細胞的多模態表示接近,同一細胞類型的表示聚在一起。
常用方法:
- Seurat v4:擴展了 Seurat v3 的 CCA 和 MNN 方法,支持多模態和多樣本整合,通過加權最近鄰(Weighted Nearest Neighbor, WNN)生成統一表示。
- Liger:用整合非負矩陣分解(integrative NMF)同時處理多模態和多樣本數據,提取共享因子。
- MOFA+:多組學因子分析,建模多模態和多樣本的共享變異。
- scVI/scANVI:用深度學習的變分自編碼器(VAE),同時校正批次效應和模態差異,適合非配對數據。
- MinNet(Lu et al., 2023):雖然主要為非配對垂直整合設計,但可以擴展到對角整合,通過孿生網絡和圖基對比損失處理多樣本、多模態數據。
例子講解:
假設你在研究癌癥,想整合兩個患者的腫瘤微環境數據,每個患者有 scRNA-seq(基因表達)和 scATAC-seq(染色質開放性)數據。你用 Seurat v4 做對角整合:
- 讀數據:加載患者 1 和患者 2 的 scRNA-seq 和 scATAC-seq 矩陣。
- 預處理:對 scRNA-seq 做對數轉換,選 2000 個高變異基因;對 scATAC-seq 轉為基因活性評分,選高變異峰值。
- 找錨點:用 WNN 算法找患者間和模態間的錨點(比如患者 1 和患者 2 的 T 細胞對應,同一患者的 scRNA-seq 和 scATAC-seq 對應)。
- 整合:把所有數據投影到統一 PCA 空間,校正批次效應和模態差異。
- 可視化:用 UMAP 畫嵌入空間,看到患者 1 和患者 2 的 T 細胞、腫瘤細胞等聚成相同簇。
- 分析:比較兩患者 T 細胞的基因表達和調控峰值,找癌癥特異性調控網絡。
結果是:你得到一張 UMAP 圖,患者 1 和患者 2 的 T 細胞聚在一起,腫瘤細胞聚在一起,每個細胞的基因表達和染色質開放性表示也對齊了。你可以進一步分析 T 細胞在癌癥中的免疫抑制機制。
應用場景
- 疾病研究:整合多個患者的 scRNA-seq 和 scATAC-seq 數據,研究疾病(如癌癥、COVID-19)中細胞類型和調控網絡的變化。比如,比較健康和重癥患者的免疫細胞多組學特征。
- 跨物種分析:整合人和小鼠的多組學數據(比如 scRNA-seq 和 scATAC-seq),找保守的細胞類型和調控機制。
- 多中心協作:整合不同實驗室的多模態數據(比如 10X Multiome 數據),生成統一的細胞圖譜,服務于大項目如人類細胞圖譜(Human Cell Atlas)。
- 動態過程研究:整合不同時間點或治療階段的多組學數據,研究細胞狀態的動態變化,比如癌癥治療前后腫瘤微環境的變化。
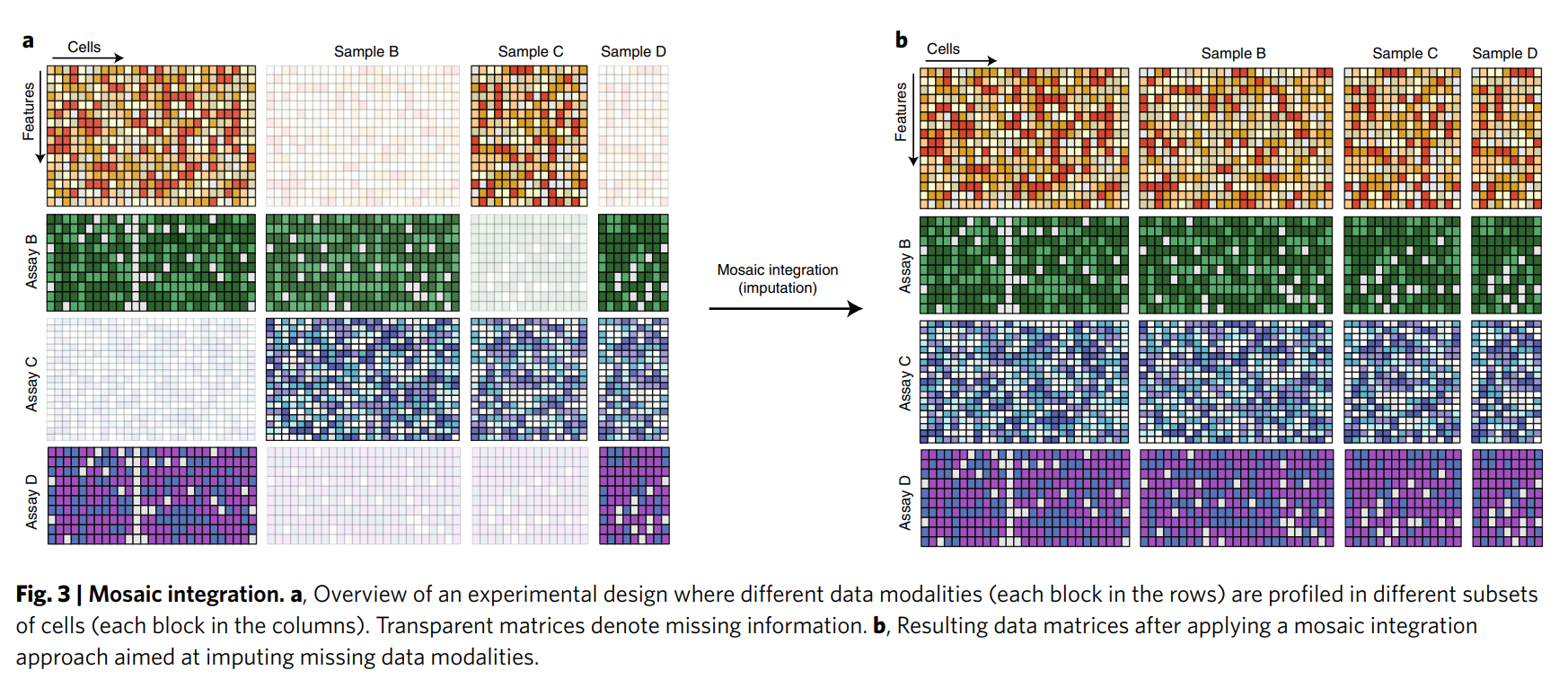
馬賽克整合(Mosaic Integration):如圖3所示,馬賽克整合涉及在不同子集上測量不同模態的數據(例如,某些細胞測RNA,另一些測蛋白質),需要通過計算方法推斷缺失數據或構建統一的表示。
?
馬賽克整合(Mosaic Integration)的目標是整合部分重疊的多樣本、多模態單細胞數據,生成一個統一的低維嵌入空間(co-embedding space)。在這個空間里:
- 同一細胞的多模態數據(比如 scRNA-seq 和 scATAC-seq)應該盡可能接近(垂直整合的目標)。
- 不同樣本的相同細胞類型(比如不同患者的 T 細胞)應該聚在一起(水平整合的目標)。
- 即使某些樣本只有單一模態數據,也能通過配對數據的“橋梁”投影到統一空間。
馬賽克整合就像拼一幅馬賽克畫,有些地方有完整的彩色和黑白圖案(配對數據),有些地方只有彩色或黑白(單一模態數據)。你用完整的圖案作為模板,把所有碎片拼成一幅畫,確保圖案對齊,顏色協調。
工作原理
- 輸入數據:
- 多樣本、多模態數據,部分樣本有配對數據(比如 Multiome 的 scRNA-seq 和 scATAC-seq),部分樣本只有單一模態(比如只有 scRNA-seq 或 scATAC-seq)。
- 數據通常是矩陣形式(行是基因/峰值,列是細胞),但不同樣本的模態組合不同。
- 預處理:
- 標準化:對每種模態數據歸一化(比如 scRNA-seq 對數轉換,scATAC-seq 轉為基因活性評分),統一統計特性。
- 特征選擇:選高變異基因(scRNA-seq)或高變異峰值(scATAC-seq),減少噪音。
- 批次標注:給每個樣本和模態標記標簽(比如患者 1 的 scRNA-seq、患者 2 的 scATAC-seq)。
- 整合方法:
- 找錨點:利用配對數據(比如患者 1 的 Multiome 數據)作為“橋梁”,找到模態間和樣本間的對應關系。比如,通過互近鄰(MNN)或典型相關分析(CCA)匹配患者 1 的 T 細胞 scRNA-seq 和 scATAC-seq,再匹配患者 1 和患者 2 的 T 細胞 scRNA-seq。
- 投影到共享空間:將所有數據(配對和非配對)映射到一個統一的低維空間,配對數據作為錨點引導非配對數據的投影。
- 校正效應:同時消除批次效應(樣本間差異)和模態效應(數據類型差異)。
- 優化目標:
- 模態對齊:最小化配對數據中同一細胞的模態間距離(比如用歐幾里得距離或 KL 散度)。
- 樣本對齊:最小化不同樣本的相同細胞類型的距離,確保 T 細胞、B 細胞等聚類一致。
- 非配對數據投影:用配對數據的錨點,推斷非配對數據的嵌入位置。
- 生物學信號保留:最大化細胞類型或狀態的分離(比如用分類損失確保細胞類型分開)。
- 輸出:一個統一的低維嵌入空間,所有細胞(無論來自哪個樣本或模態)都表示為點,配對數據的模態表示接近,同一細胞類型的表示聚在一起,非配對數據通過錨點合理定位。
常用方法:
- Seurat v4:用加權最近鄰(Weighted Nearest Neighbor, WNN)方法,通過配對數據作為錨點,整合多樣本、多模態數據。
- Liger:用整合非負矩陣分解(integrative NMF),利用配對數據提取共享因子,投影非配對數據。
- MOFA+:多組學因子分析,建模配對和非配對數據的共享變異。
- scVI/scANVI:用深度學習的變分自編碼器(VAE),通過配對數據學習模態和樣本的聯合分布,推斷非配對數據的表示。
- GLUE:用圖神經網絡(GNN)整合多模態數據,適合馬賽克整合場景,利用配對數據構建跨模態的圖結構。
例子講解
假設你在研究癌癥,收集了三個患者的腫瘤微環境數據:
- 患者 1:有 scRNA-seq 和 scATAC-seq 數據(配對,Multiome 技術)。
- 患者 2:只有 scRNA-seq 數據。
- 患者 3:只有 scATAC-seq 數據。
你用 Seurat v4 做馬賽克整合:
- 讀數據:加載患者 1 的配對數據(scRNA-seq 和 scATAC-seq)、患者 2 的 scRNA-seq、患者 3 的 scATAC-seq。
- 預處理:對 scRNA-seq 做對數轉換,選 2000 個高變異基因;對 scATAC-seq 轉為基因活性評分,選高變異峰值。
- 找錨點:用 WNN 算法,先匹配患者 1 的 scRNA-seq 和 scATAC-seq(模態間錨點),再匹配患者 1 和患者 2 的 scRNA-seq(樣本間錨點),以及患者 1 和患者 3 的 scATAC-seq。
- 整合:把所有數據投影到統一 PCA 空間,用患者 1 的配對數據作為橋梁,校正批次和模態效應。
- 可視化:用 UMAP 畫嵌入空間,看到患者 1、2、3 的 T 細胞、腫瘤細胞等聚成相同簇,患者 1 的 scRNA-seq 和 scATAC-seq 表示對齊。
- 分析:比較三患者 T 細胞的基因表達和調控峰值,找癌癥特異性調控網絡。
結果是:你得到一張 UMAP 圖,三患者的 T 細胞聚在一起,患者 1 的基因表達和染色質開放性對齊,患者 2 和 3 的單一模態數據也合理定位。你可以分析 T 細胞在癌癥中的免疫抑制機制。
應用場景
- 疾病研究:整合多患者的部分多組學數據,研究疾病機制。比如,整合 COVID-19 患者的配對和非配對 scRNA-seq、scATAC-seq 數據,比較免疫細胞的分子特征。
- 跨物種分析:整合人和小鼠的部分多組學數據(比如有些樣本只有 scRNA-seq,有些有 scRNA-seq 和 scATAC-seq),找保守的細胞類型和調控機制。
- 多中心協作:整合不同實驗室的部分多組學數據,生成統一細胞圖譜。比如,人類細胞圖譜(Human Cell Atlas)可能只有部分樣本有配對數據。
- 動態過程研究:整合不同時間點或治療階段的部分多組學數據,研究細胞狀態變化,比如癌癥治療前后腫瘤微環境的變化。
3. 數據整合的統計挑戰
文檔中的Box 1詳細列出了單細胞多組學分析的統計挑戰,文章可能深入探討了以下問題:

- 異質性數據模態:不同實驗技術(如scRNA-seq、scNMT-seq、scCOMT-seq)生成的數據具有不同的統計特性。例如,RNA-seq測量mRNA表達,而DNA甲基化數據基于CpG位點,這些數據需要統一的統計框架來整合,但結合不同似然模型(likelihood models)是一項復雜任務。
- 過擬合(Overfitting):單細胞數據通常涉及高維特征(例如,數百萬個CpG位點),而樣本量有限(僅幾百個細胞),導致過擬合風險。文章提到正則化和降維技術(如PCA或變分自編碼器VAE)是應對這一問題的關鍵。
- 缺失數據:不同技術對“缺失數據”的定義不同。例如,scNMT-seq不區分基因組特征的“存在”與“缺失”,而scCOMT-seq需要考慮實驗設計的缺失信息。文章可能討論了如何通過統計方法(如線性回歸、PCA或VAE)處理缺失數據。
- 混雜因素:某些模態特有的混雜因素(如全局甲基化水平受組蛋白修飾影響)可能干擾整合結果。文章可能提出通過添加協變量或使用匹配的負對照來解決此類問題。
4. 數據整合的應用
文章可能通過具體案例展示了數據整合的應用,例如:
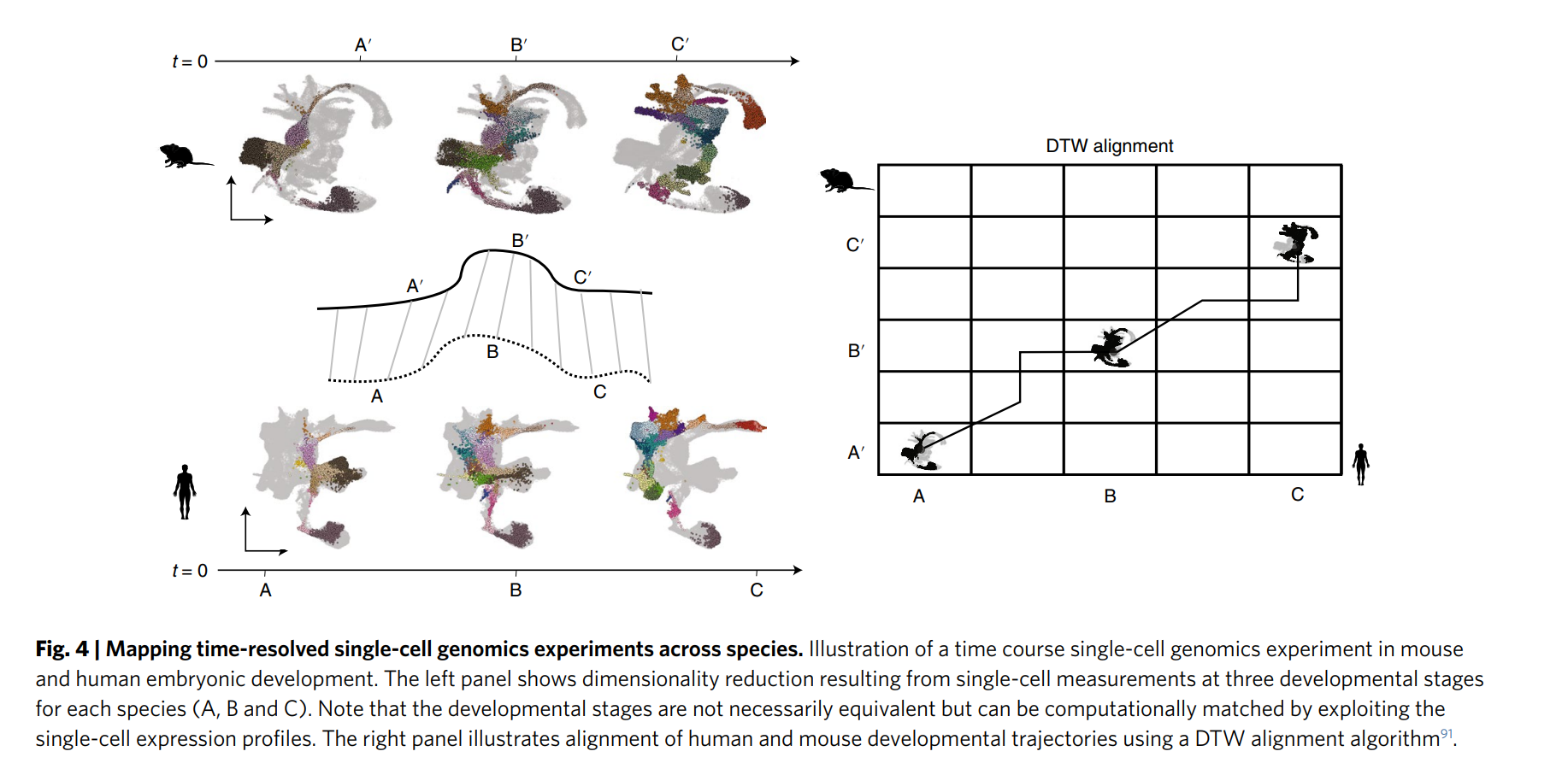
- 跨物種比較:如圖4所示,通過時間分辨的單細胞基因組學實驗,比較鼠和人胚胎發育的細胞狀態,揭示物種間的保守性和差異性。降維技術(如t-SNE或UMAP)被用于可視化這些數據。???????
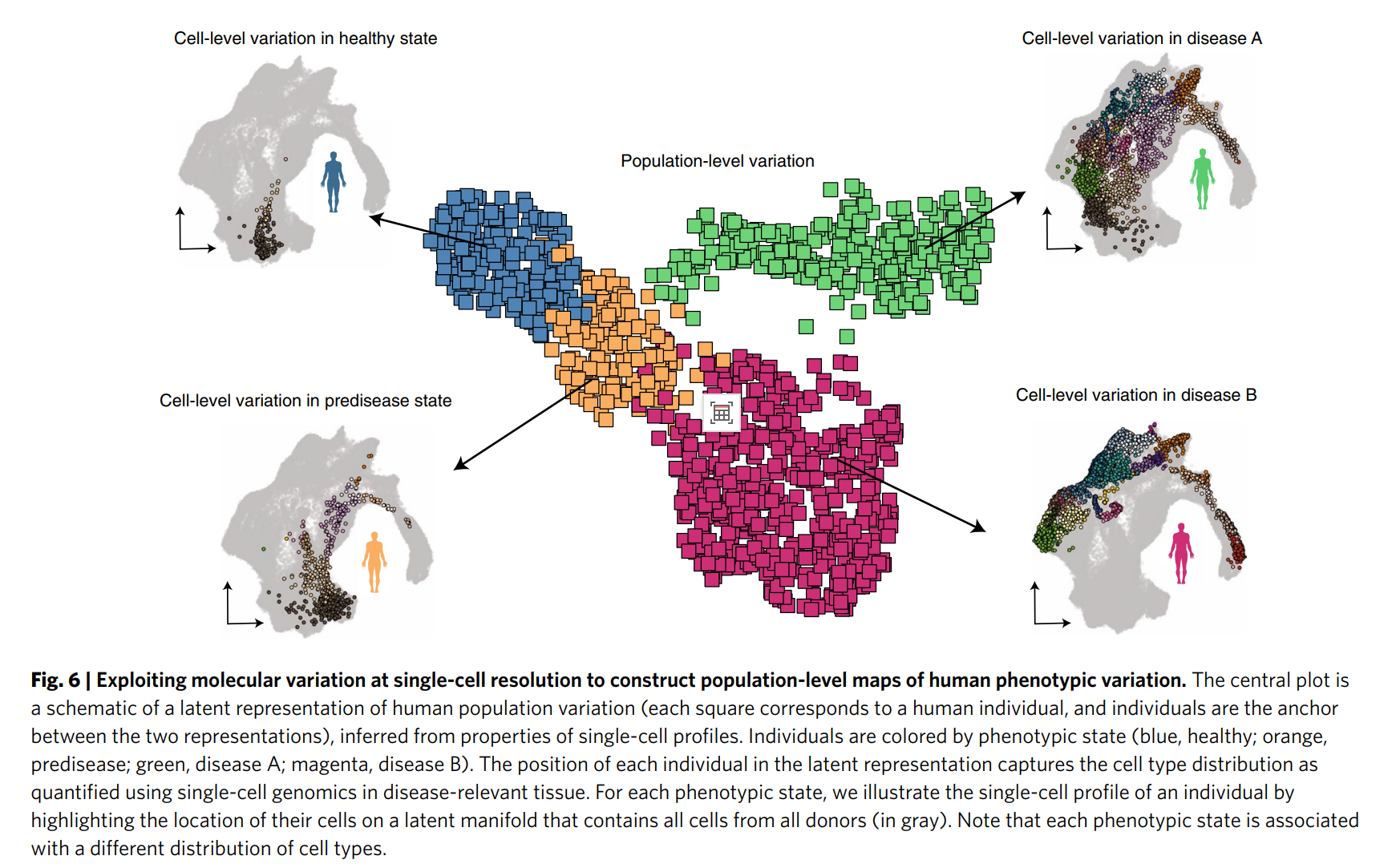
- 疾病研究:如圖6和頁面12所述,單細胞數據整合可用于構建健康和疾病狀態下的人類表型變異圖譜。例如,通過分析13種癌癥患者的免疫細胞組成,進行患者分層,助力精準醫學。
- 多尺度建模:頁面12提到通過模型驅動或專家知識定義特征,用于提取細胞表示并進行患者分層。這類方法在未來可能推動多組學數據與臨床表型的關聯研究。
5. 數據集與方法評估
文檔中的Table 2提到了一些用于基準測試(benchmarking)的數據集,涵蓋水平、垂直、對角和馬賽克整合任務。這些數據集可能包括:
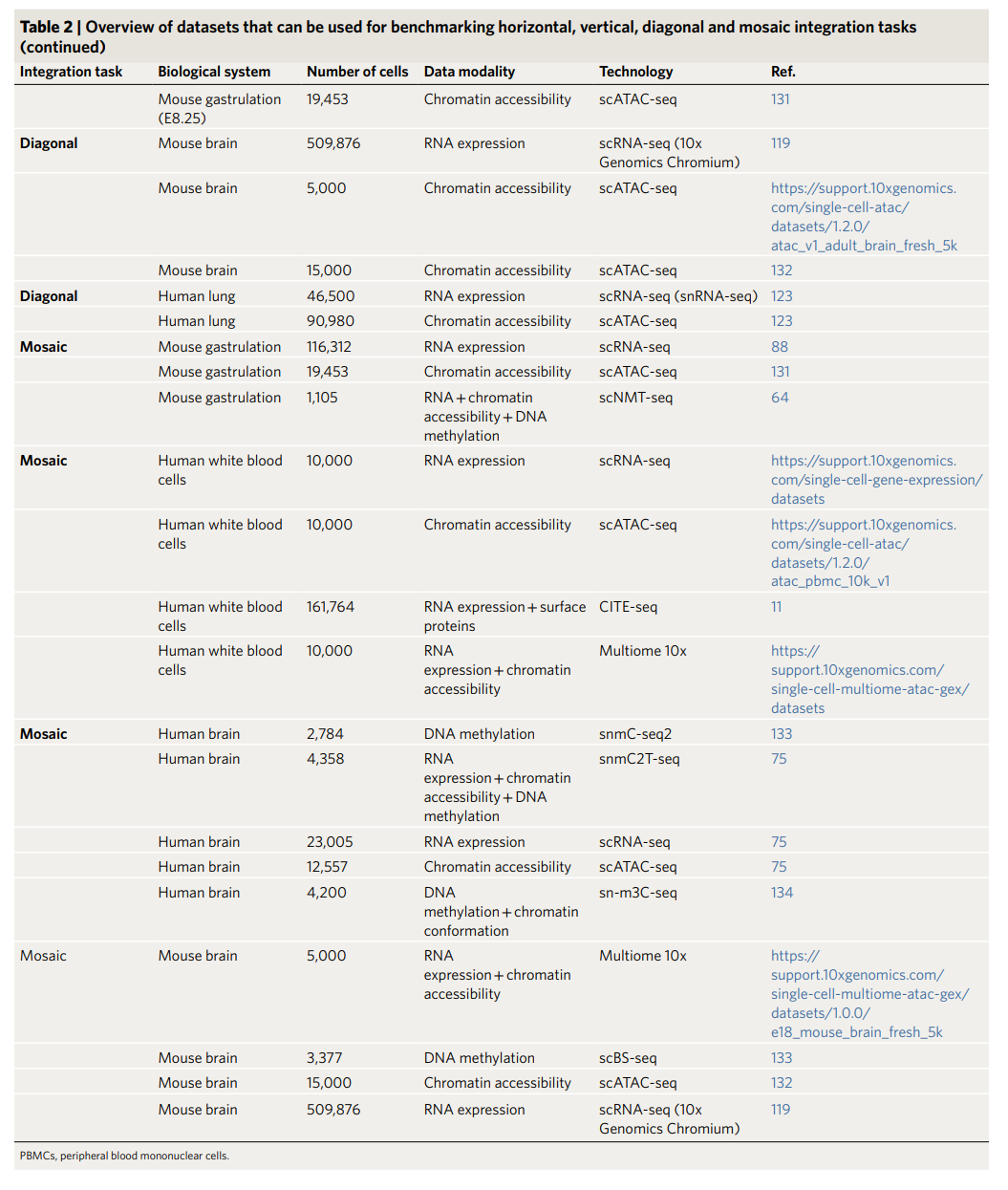
- 生物系統:例如,免疫細胞、胚胎發育、癌癥組織等。
- 數據模態:包括RNA表達、表觀遺傳數據、蛋白質分布等。
- 技術:如scRNA-seq、scATAC-seq等單細胞技術。
- 細胞數量:不同數據集的細胞數量差異,用于評估整合方法的擴展性。
6. 整合類型對比
| 特性 | 馬賽克整合 | 對角整合 | 水平整合 | 垂直整合 |
|---|---|---|---|---|
| 目標 | 整合部分配對的多樣本、多模態數據,消除批次和模態效應 | 整合多樣本、多模態數據,消除批次和模態效應 | 整合多樣本的同模態數據,消除批次效應 | 整合同樣本的多模態數據,統一表示空間 |
| 數據類型 | 部分配對、多樣本、多模態(比如部分 scRNA-seq + scATAC-seq) | 多樣本、多模態(比如 scRNA-seq + scATAC-seq) | 同模態(比如全是 scRNA-seq) | 多模態(比如 scRNA-seq + scATAC-seq) |
| 應用場景 | 不完整數據集整合、跨患者多組學、跨物種分析 | 跨患者多組學比較、跨物種調控分析 | 跨樣本/患者比較、跨物種分析 | 多組學細胞圖譜、調控網絡推斷 |
| 主要挑戰 | 數據不完整、雙重異質性、錨點選擇、高維數據 | 雙重異質性、錨點選擇、高維數據、過擬合 | 批次效應、錨點選擇、生物學變異保留 | 模態異質性、高維數據、缺失數據 |
| 常用方法 | Seurat v4 (WNN)、Liger、MOFA+、scVI、GLUE | Seurat v4 (WNN)、Liger、MOFA+、scVI | Seurat v3 (CCA/MNN)、Harmony、Liger | Seurat v3 (CCA)、Liger、MOFA+、MinNet |
| 輸出 | 統一嵌入空間,模態和樣本對齊,非配對數據投影 | 統一嵌入空間,模態和樣本對齊 | 統一嵌入空間,相同細胞類型對齊 | 統一嵌入空間,同一細胞的多模態表示對齊 |
通俗比喻:
- 馬賽克整合:拼一幅馬賽克畫,有些地方有彩色和黑白圖案(配對數據),有些只有彩色或黑白(非配對數據),用完整圖案橋接所有碎片。
- 對角整合:拼不同國家攝影師的彩色和黑白照片,每國都有兩種照片。
- 水平整合:拼不同國家攝影師的彩色照片,確保“山”和“樹”對齊。
- 垂直整合:拼一個國家攝影師的彩色和黑白照片,確保同一個物體重合。
?









)




)



)
)