生信堿移
貝葉斯網絡聚類
CANclust是一種基于貝葉斯的聚類方法,系統性地對基因突變、細胞遺傳學信息和臨床指標進行聯合建模,用于多種模態數據的聯合聚類分析,并識別在患者群體中反復出現的特征模式。
個體的遺傳與環境背景決定其應對疾病的反應狀態,進而產生單一疾病的不同疾病分型。不同疾病分型的病人具有高度異質性,但在臨床表現和基因特征上可能存在顯著重疊。比如,髓系腫瘤包括急性髓系白血病(AML)、骨髓增生異常綜合征(MDS)、慢性髓單核細胞白血病(CMML)和骨髓增殖性腫瘤(MPN)等多個疾病類型,他們之間并非截然分離,而是構成一個連續譜系,其中MDS、CMML 和 MPN 等可進一步進展為AML。
盡管近年來臨床分型方法已經逐步由傳統臨床指標向突變特征轉變,但當前分類體系仍面臨三大問題:
-
異質性顯著:即使是同一疾病,患者在臨床表現、預后以及治療響應方面存在廣泛的差異;
-
變量組合十分復雜:基因組層面驅動突變數量眾多、相互組合多樣,臨床協變量(如血液、骨髓指標)與遺傳因素的聯合分析復雜度極高;
-
聚類方法有限:多數已有聚類研究僅關注基因層面,忽略了臨床協變量在亞型識別中的重要性,同時現有的方法在建模中無法體現不同變量類型的差異角色。
為此,來自瑞士生物信息學研究所的研究人員開發了一種基于貝葉斯網絡的協變量校正聚類方法(CANclust),于2025年4月30日發表于Nature Communications[IF: 14.7]。CANclust從數據整體視角出發,系統性地對基因突變、細胞遺傳學信息和臨床指標進行聯合建模,用于多種模態數據的聯合聚類分析。作者在正文中利用模擬、TCGA、自測數據進行了驗證分析,不過像聚類結果出來以后一些套路性的分析比如免疫治療那些都可以做起來了

▲ DOI:10.1038/s41467-025-59374-1。?
簡單來講,CANclust通過建模變量之間的概率關系,識別在患者群體中反復出現的特征模式。對于每一個潛在的患者亞型(聚類結果),該方法都會用一個貝葉斯網絡來描述變量間的特征組合,所以不僅考慮了單個特征,也關注了特征變量之間的協同變化。不理解也沒關系,反正使用這個聚類方法我們可以除了獲得樣本聚類結果以外,還能夠獲得不同聚類特異性的特征交互網絡。
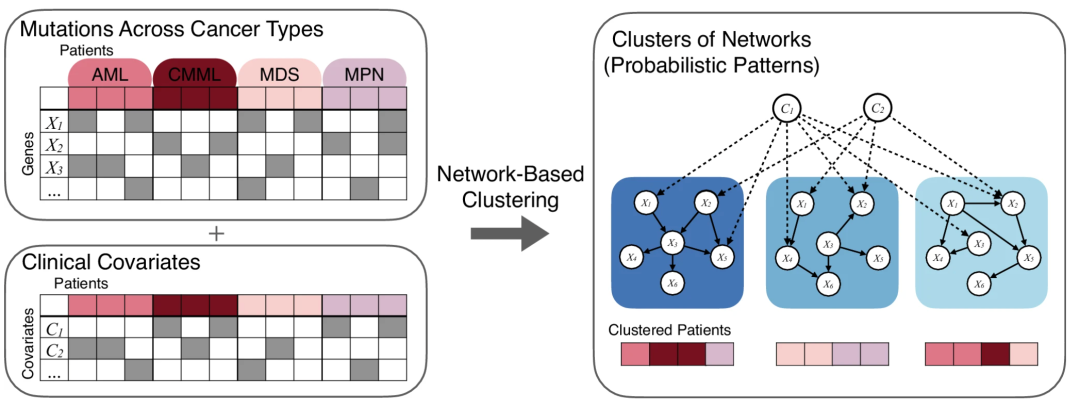
▲ CANcluster原理。該方法以患者在確診時的基線數據為輸入(圖左),包括不同癌癥類型(左側顏色區分)、基因突變信息(左上)和臨床協變量(左下)。在建模過程中,通過區分協變量類型:Cluster-independent 協變量(如年齡、性別)被設定為僅向突變變量發出邊(圖右虛線),用于建模其對突變發生的影響,并在聚類時進行統計校正;Cluster-dependent 協變量(如癌癥類型)則被視為可能由突變決定的結果變量,可與突變變量共同參與聚類建模。隨后貝葉斯網絡對不同潛在亞型中的變量依賴結構進行建模(右側藍色圖結構),每個亞型對應一個特有的網絡模型,反映其內在的突變模式與協變量關聯結構(圖右實線)。通過聯合學習網絡結構與患者歸屬(圖右下),該方法能夠在控制已知協變量影響的基礎上,識別出具有生物學一致性和臨床解釋力的全新患者亞群。
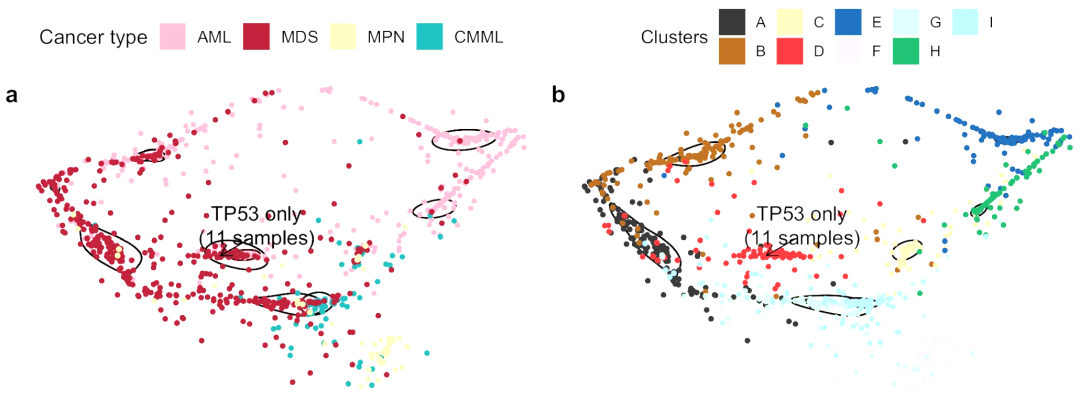
▲ 樣本聚類結果演示。一個點代表一個樣本,左圖為樣本真實的癌癥類型;右圖為對應的聚類結果,總共有A,B,C,D,E,F,G,H,I九個聚類簇。上圖為作者NC文章中的結果,表明該方法能夠發現新的臨床亞型。
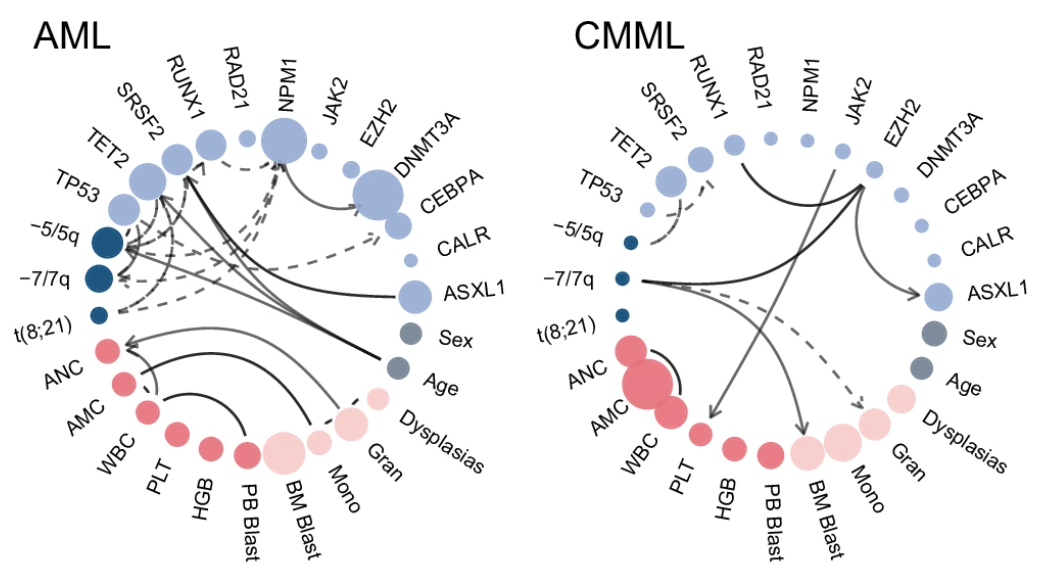
▲ 兩種癌癥中的特異性網絡。除了突變特征以外,還有性別、年齡等臨床信息。實線代表正相關,而虛線代表負相關。
01.R包安裝
該R包在CRAN中提供,可以使用以下代碼進行安裝:
install.packages("clustNet")
02.使用示例
①示例數據加載:
library(clustNet)# 模擬數據
ss <- c(400,?500,?600)?# samples in each cluster
simulation_data <- sampleData(k_clust = k_clust, n_vars =?20, n_samples = ss)
sampled_data <- simulation_data$sampled_data
模擬了一個基因突變的數據,行是樣本共1500個,列是基因共20個。這個數據可以換成其它類型的數據,但是必須是0或1的類別變量。下面簡單來看一下:
dim(sampled_data)
#[1] 1500 ? 20# 因為是基因突變數據,所以0/1分別代表無或有突變
sampled_data[1:5,?1:5][,1] [,2] [,3] [,4] [,5]
[1,] ? ?1? ??0? ??0? ??0? ??1
[2,] ? ?0? ??1? ??1? ??1? ??0
[3,] ? ?1? ??0? ??0? ??0? ??0
[4,] ? ?1? ??0? ??1? ??0? ??0
[5,] ? ?0? ??0? ??1? ??0? ??0
② 使用get_clusters函數進行聚類,可以指定聚類數量(下面是指定聚為3類)。需要注意一下,sampled_data必須是0/1的變量;n_bg參數默認為0,可以用于指定需要調整的協變量如性別年齡等,但是需要放在sample_data的最后n_bg列(所以默認的0則意味著不需要考慮任何的協變量)。
k_clust <-?3?# 選定聚類為3類
cluster_results <- get_clusters(sampled_data, k_clust = k_clust, n_bg =?0)
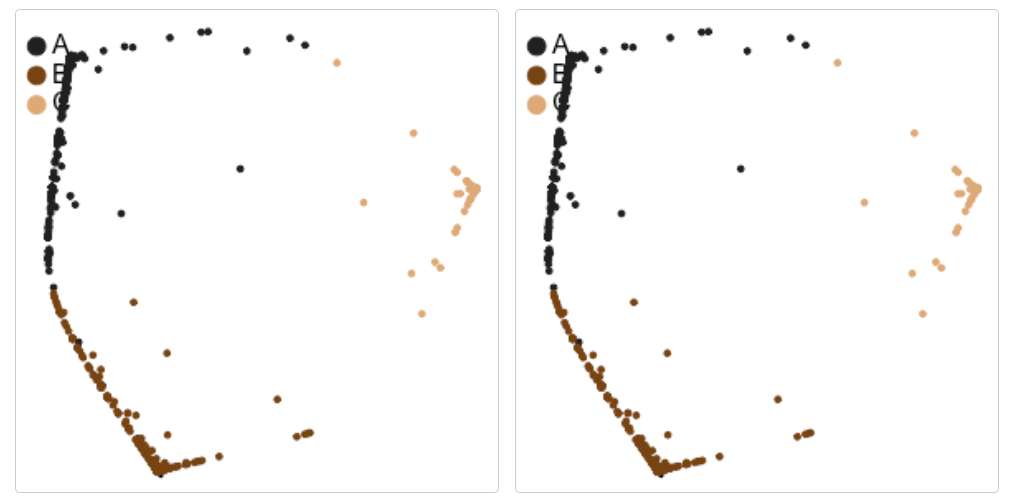
可視化一下聚類的結果圖:
library(car)
library(ks)
library(graphics)
library(stats)# 2D降維結果
density_plot(cluster_results)
③ 可視化一下聚類特異性特征網絡:
library(ggplot2)
library(ggraph)
library(igraph)
library(ggpubr)# 可視化
plot_clusters(cluster_results)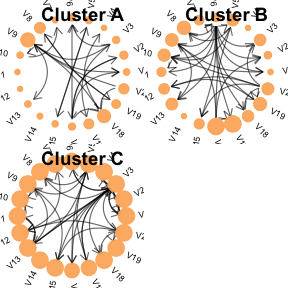
各位老鐵51快樂啊
得到工作日了










)



)




