可能出現的問題:
*個人電腦上遇到不能collect memery的情況,是電腦內存較少,建議分成用2G左右的數據進行組裝。
* Seed.fasta #用于起始組裝的種子序列,NOVOPlasty安裝軟件目錄下有這個文件,就叫這個名字,作者親測還是這個種子序列最好用。
----------------------------------------------------------------
葉綠體是絕大多數陸生植物都有的細胞器,讓人驚奇的是,植物葉綠體和線粒體一樣,都有著自己一套遺傳物質。本文介紹如何用高通量測序數據利用兩款常用的葉綠體基因組組裝軟件(NOVOPlasty和Get Organelle)來組裝完整的葉綠體基因組。
兩個軟件都是命令工行
1.軟件安裝
NOVOPlasty
1.1系統和語言安裝
NOVOPlasty是由perl語言寫的,先讓自己的電腦安裝perl語言的編譯器,
https://www.perl.org/
建議使用Linux系統,電腦已經是linux 系統或者macos的就不用管了,windows系統可以下載subsystem for windows具體的操作就不再這里描述了,可以參考下面兩個連接里面的內容。確保你的電腦是有大于等于8G的運行內存。
WSL(Windows Subsystem for Linux)的安裝與使用?www.cnblogs.com
安裝好了運行Ubuntu 18的系統
1.2 NOVOPLasty 安裝
在命令行輸入
git clone ndierckx/NOVOPlasty
下載
ndierckx/NOVOPlasty?github.com
上面的連接是NOVOPlasty在github的連接,有問題可以參考
1.3 運行NOVOPlasty
運行NOVOPlasty非常簡單
perl +NOVOPLasty的安裝路徑/NOVOPlasty.pl -c config.txt
就可以了,重點在于NOVOPlasy的配置文件也就是cofig.txt的設置
Project:
-----------------------
Project name = #你的項目名稱
Type = mito #組裝類型葉綠體chloro 或者是線粒體mito
Genome Range = 12000-22000 #基因組的預估大小,基本上在16K左右吧,找個近緣物種看看大小
K-mer = 33 #用于組裝的k-mer 的大小,并不是越大越好,一般不建議更改
Max memory = #最大運行內存
Extended log = 0
Save assembled reads = no
Seed Input = Seed.fasta #用于起始組裝的種子序列,NOVOPlasty安裝軟件目錄下有這個文件,作者親測還是這個種子序列最好用,注意寫絕對路徑。
Extend seed directly = no
Reference sequence = /path/to/reference_file/reference.fasta (optional)#選填,參考的近緣物種參考序列
Variance detection = no
Heteroplasmy = #檢測異質性的參數,不填
HP exclude list =
Chloroplast sequence = /path/to/chloroplast_file/chloroplast.fasta (only for "mito_plant" option) #組裝植物線粒體的時候填,提供本物種的葉綠體序列
Dataset 1:
-----------------------
Read Length = 151#高通量reads讀長
Insert size = 300 #插入片段大小,測序建庫的時候有
Platform = illumina #目前只支持illumina等大公司,BGI不支持注意
Single/Paired = PE #雙端測序還是單端測序
Combined reads =
Forward reads = /path/to/reads/reads_1.fastq #前reads的文件路徑
Reverse reads = /path/to/reads/reads_2.fastq #后reads 的文件路徑
Optional:
-----------------------
Insert size auto = yes
Use Quality Scores = no
直接更改config.txt文件內容就可以了。
1.4 結果解讀
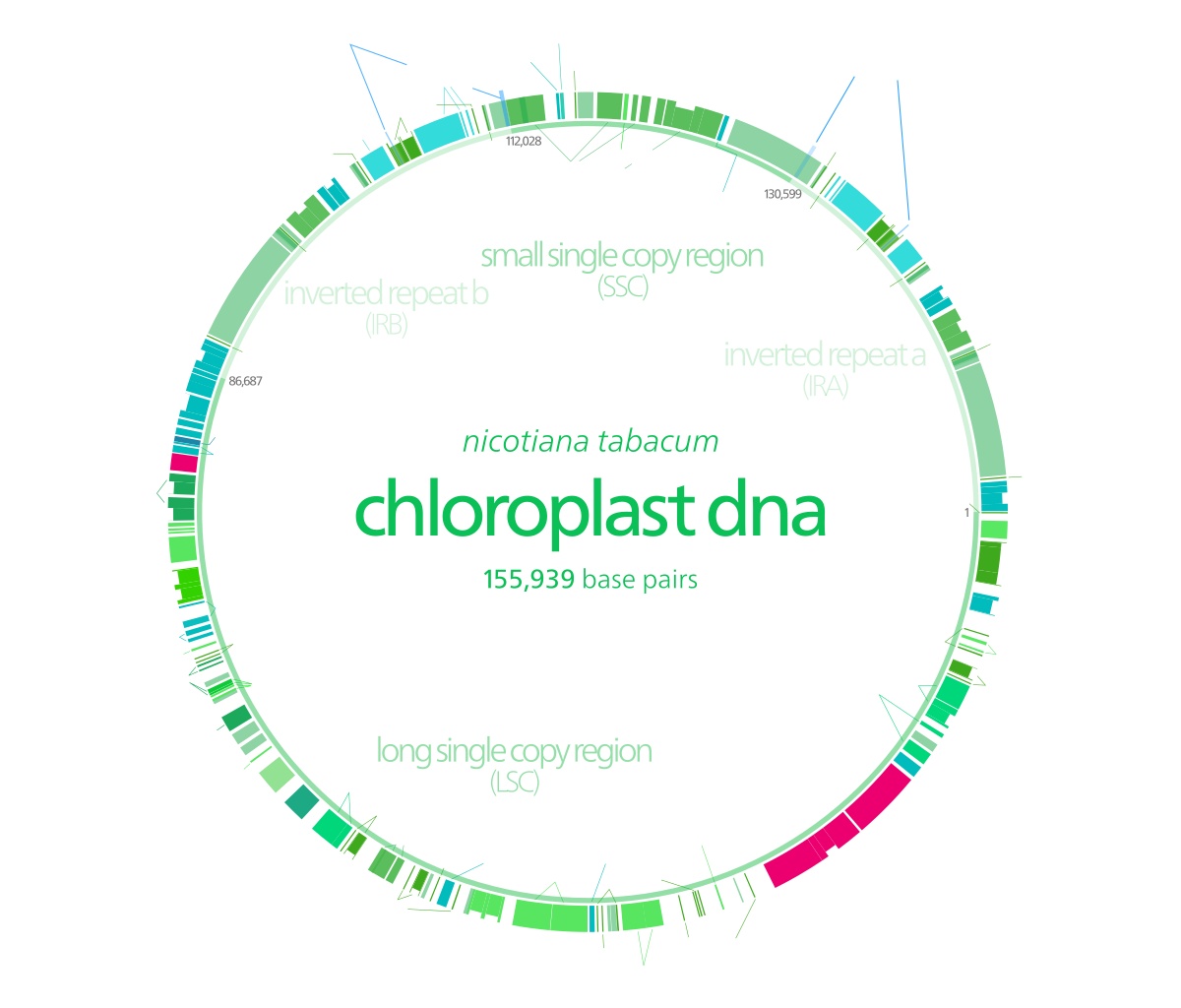
組裝質量最好會出現一個circular assembly 的文件,不過一般情況下會有兩個option,這兩個文件都是正確的,是葉綠體的兩個反向重復區域的基因順序不確定造成的。這個時候想確定哪一個正確的時候,就要做基因注釋或者PCR驗證。
還有的時候文件會出現*的堿基,這個時候也是軟件不能處理一些ambiguous的區域,需要人為將兩端的序列比對一下,去掉*號,我組裝的結果都沒有出現這樣的情況,但是對于結果也檢查一下有無這種情況出現。
組裝質量差的情況下,會出下多個option組裝結果,或者僅有多個contigs,這個時候比較麻煩了,就要選擇reference genome重新組裝或者更換其他的軟件,getorganelle也是用于組裝葉綠體的軟件,據這個軟件作者來說,相較于NOVOPlasty有更高的準確性,NOVOplasty速度快,使用簡單,能夠滿足大多數的需求。有時間再把getorganelle軟件的用法寫一寫。
后期檢查組裝結果的話建議對基因組進行注釋,軟件推薦Geseq基因注釋 rna注釋,可視化真的是一鍵到位,操作也是非常傻瓜,有需要的話給大家介紹。


)







慢得多.為什么?)







)
)