VET:Vcf Export Tools
工具簡介
VET是一個基于R語言開發的變異位點信息批量提取工具,主要功能是根據VCF數據集,按照基因ID、樣品ID、變異位點ID等參數,實現批量提取,同時支持變異位點結構注釋,一步搞定變異數據的快速提取。
##########?WelCome?to?VCF?Export?Tools?###########
>>>>>>>>>>>>>>>>?Design?By?BioNote?<<<<<<<<<<<<<<<<<
可選參數:
[1]根據基因ID提取變異數據
[2]根據物理位置提取變異數據
[3]根據樣品名稱提取變異數據
[4]根據SNP名稱提取變異數據
--------------------------------------------------
[INFO]第一個參數填選項,第二個參數填項目備注名稱
[INFO]第三個參數選擇是否過濾樣本,默認為“N”
[INFO]第四個參數選擇是否進行結構注釋,默認為“N”
[INFO]運行方法?$?Rscript?./run.R?1?test?Y?Y
>>>>>>>>>>>>>>>>?程序版本:V?2.0.1?<<<<<<<<<<<<<<<<
##################################################
功能與應用
基因測序后經過上游分析得到的VCF文件儲存了所有樣本對應的所有變異信息,通常數據量非常大,在實際使用中需要根據情況對指定信息進行提取。目前已有vcftools或bcftools等工具能夠實現上述操作,但是用的時候參數比較復雜,整個過程略顯繁瑣。
本工具集成了R、tidyverse、Python、vcftools、bcftools、snpEff等常用工具,開發便捷式流程實現批量操作。
主要應用場景是對大規模VCF原始數據集進行提取,支持多種個性化方式:
-
按基因ID提取指定基因內變異信息 -
按材料名稱提取某些材料變異信息 -
按變異位點名稱篩選指定變異信息 -
按照物理位置提取指定區段內數據
支持流式操作,提取后篩選指定樣品并對每個變異位點進行結構注釋(判斷錯義突變、移碼突變等),最終將結果文件打包生成壓縮包。
使用方法
-
第一步:輸入待提取的信息 -
第二步:運行Run.R腳本 -
第三步:下載結果文件
可以批量操作,無需手寫代碼。
原理介紹
VCF是生信研究中儲存基因變異信息的重要格式,通常經過測序上游分析后得到一份具有豐富信息量的vcf或者vcf.gz文件。

以“##”開頭的行表示注釋信息,一般記錄了字段類型和歷史命令,這部分相當于一個日志信息。剩下的數據部分類似一個表格,大體上每行是一個變異位點,每列是一個材料樣本。
變異位點(簡稱SNP)是按照染色體上不同位置進行統計的,展示不同材料中在某個位置堿基差異。
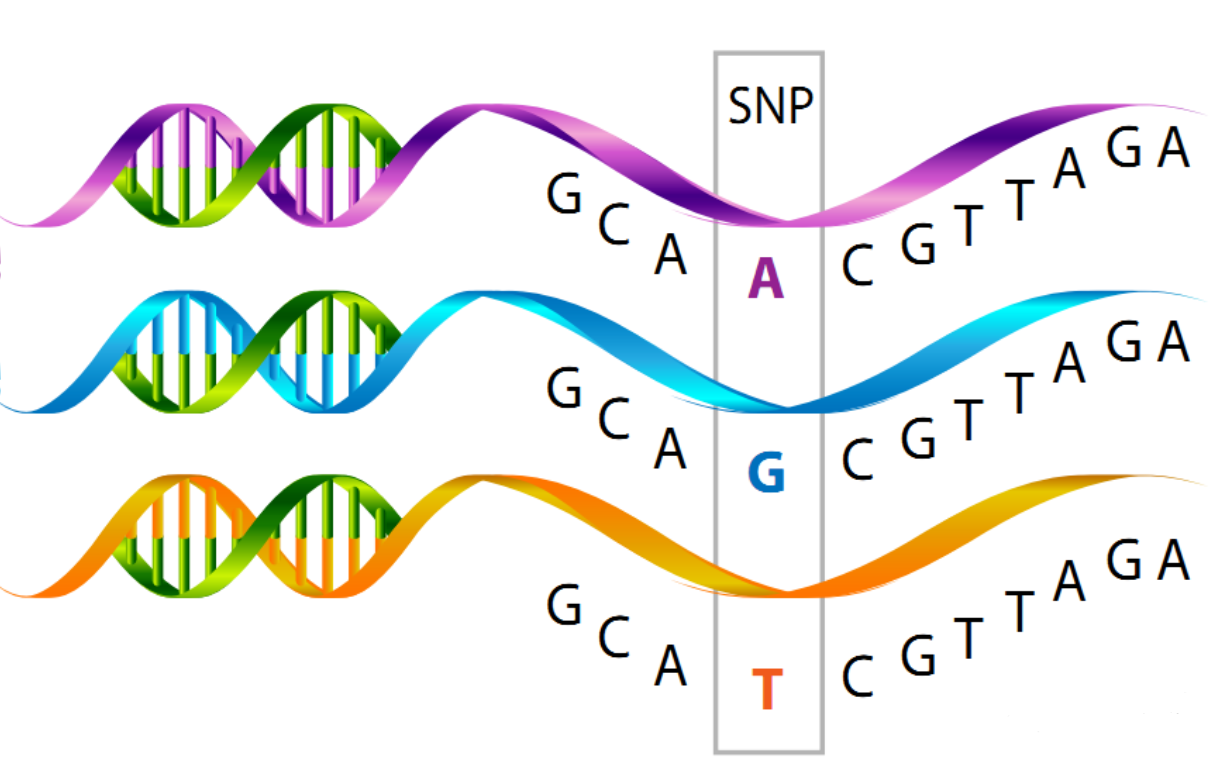
按照突變的類型可以分為3種類型:
-
缺失:Del,某些堿基不見了 -
插入:Ins,新出現了某些堿基 -
替換:SNP,單核苷酸多態性變異
 一般常見的VCF文件主要由上述信息組成,對于大規模測序得到的多個樣品合并VCF文件,可能包含幾千萬行×幾千列(億級數據量)。
一般常見的VCF文件主要由上述信息組成,對于大規模測序得到的多個樣品合并VCF文件,可能包含幾千萬行×幾千列(億級數據量)。
在實際進行分析時,可能只需要考慮某幾個基因或者是一小段區間內的變異數據,因此需要對VCF文件進行提取,只取出想要的一小部分,這個過程涉及到Linux下不同軟件的相互配合。
VET 源代碼
首先,建立項目文件夾并生成以下結構:
Aug??9?16:30?00_scripts
Aug?17?15:24?01_INPUT_GeneID.txt
Aug?17?16:09?01_out_byGeneID
Aug??9?18:30?02_INPUT_Postion.txt
Aug?10?11:25?02_out_byPostion
Aug?10?11:02?03_INPUT_SampleName.txt
Aug??4?15:13?03_out_bySampleName
Aug??4?15:31?04_INPUT_SNP.txt
Aug??4?15:14?04_out_bySNP
Aug??6?11:34?05_INPUT_filevcf.txt
Aug??6?11:33?05_out_bySnpEff
程序初始化
下面的代碼為程序初始化過程,將會加載tidyverse等軟件包,并讀取重要參數,完成后將獲得輸出提示。
#!/usr/local/bin/Rscript
#?VCF?Export?Tools?基因型變異數據批量提取工具,快捷提取VCF文件
#?依賴軟件:Python、bcftools、tidyverse、snpeff
suppressPackageStartupMessages(library("cli"))
suppressPackageStartupMessages(library("tidyverse"))
cli::cli_text("##########?WelCome?to?VCF?Export?Tools?###########
?\n?>>>>>>>>>>>>>>>>?Design?By?Jewel?<<<<<<<<<<<<<<<<<
?\n可選參數:
?\n\t[1]根據基因ID提取變異數據
?\n\t[2]根據物理位置提取變異數據
?\n\t[3]根據樣品名稱提取變異數據
?\n\t[4]根據SNP名稱提取變異數據
?\n--------------------------------------------------
?\n[INFO]第一個參數填選項,第二個參數填項目備注名稱
?\n[INFO]第三個參數選擇是否過濾樣本,Y為過濾指定樣本
?\n[INFO]第四個參數為'Y'時將對vcf文件進行變異結構注釋
?\n[INFO]例如?$?./run.R?1?test?Y?Y
?\n>>>>>>>>>>>>>>>>?程序版本:V?2.0.1?<<<<<<<<<<<<<<<<
?\n?##################################################")
?args?<-?commandArgs(T)
if(length(args)!=4){stop("參數輸入有誤,請檢查輸入格式,示例“./Run.R?1?jobname?Y/N?Y/N")}
#?CONFIG?SETTING:
db_file?<-?"wgs_all.vcf.gz"?#?設置數據庫名稱
db_name?<-?"WGS"
#?程序初始化,刪除上次輸出結果文件----------
OPT?<-?args[1]?#?程序子選項
JOB?<-?args[2]?#?項目備注信息
SAM?<-?args[3]?#?是否過濾樣本
EFF?<-?args[4]?#?是否結構注釋
print(str_c("INFO???當前選擇的數據庫為:",db_file))
print(str_c("INFO???當前項目名稱為:?",OPT,"?<->?",JOB))
print(str_c("INFO???是否對樣品進行過濾(Y為過濾指定樣本,否則不過濾):"),SAM)
print(str_c("INFO???是否對vcf進行結構變異注釋(Y為進行注釋,否則不注釋):"),EFF)
system("rm?-rf?./01_out_byGeneID/*")
system("rm?-rf?./02_out_byPostion/*")
system("rm?-rf?./03_out_bySampleName/*")
system("rm?-rf?./04_out_bySNP/*")
system("rm?-rf?./05_out_bySnpEff/*")
cli::cli_text("INFO???系統輸出文件夾初始化完成")
根據基因ID提取變異信息
根據輸入的參數進行判斷,如果選項為1,則執行下面的步驟,主要調用Python程序進行信息檢索,并由bcftools工具批量提取變異信息,若需要根據指定樣品進行過濾,則利用view功能對樣品進行篩選,最后生成結果壓縮文件。
if?(OPT?==?"1"){
??cli::cli_text("INFO???待提取的基因ID如下,將自動自取上下游3000bp內的變異數據")
??id?<-?read.table("./01_INPUT_GeneID.txt",header?=?F)
??print(id$V1)
??cli::cli_text("INFO???基因ID信息整理完畢,接下來開始檢索物理區間")
??system("Rscript?prefix_gene_filter.R?./01_INPUT_GeneID.txt")
??cli::cli_text("INFO???接下來執行Python腳本調用bcftools提取基因變異信息")
??system(str_c("python?bcftools_view_filiter_Chr.py?--input?./00_scripts/id.txt?--vcf?./",
???????????????db_file))
??cli::cli_text("INFO???提取完成,對結果進行打包壓縮")
??
??if?(SAM?==?"Y"){
????for?(i?in?1:nrow(id)){
??????system(str_c("bcftools?view?--force-samples?-S?",
???????????????????"./03_INPUT_SampleName.txt?",
???????????????????id$V1[i],".vcf.gz?>?",
???????????????????id$V1[i],".vcf"))
????}
????system("mv?./Traes*vcf?./01_out_byGeneID/")
????system("rm?-rf?./Traes*vcf.gz")
????system(str_c("tar?-czvf?",format(Sys.Date(),?"%Y_%m_%d"),"_",JOB,"_ExportFrom_",db_name,
?????????????????"_LOTSample_Filter_ByGeneID",".tar.gz?./01_out_byGeneID/*?./Tips.pdf"))
??}else{
????system("mv?./Traes*?./01_out_byGeneID/")
????system(str_c("tar?-czvf?",format(Sys.Date(),?"%Y_%m_%d"),"_",JOB,"_ExportFrom_",db_name,
?????????????????"_AllSample_Filter_ByGeneID",".tar.gz?./01_out_byGeneID/*?./Tips.pdf"))
??}
??cli::cli_text("INFO???任務運行結束,請及時下載結果文件,下次運行前將清空結果文件")
}
根據物理位置提取變異信息
根據指定的物理區間判斷染色體的和起止位置,并結果VCF文件篩選指定區間內的變異數據,采用bcftools的 filter功能進行實現,提取完成后進行打包壓縮。
if?(OPT?==?"2"){
??cli::cli_text("INFO???待提取物理區間如下,正在提取中......")
??region?<-?read.table("./02_INPUT_Postion.txt",header?=?F)
??for?(i?in?1:nrow(region)){
????print(str_c("Index:?",i,"???Region:?",region$V1[i],"???Info:?",region$V2[i]))
????system(str_c("bcftools?filter?",db_file,"?--regions?",region$V1[i],"?>?",region$V1[i],"_",region$V2[i],".vcf"))
????system("mv?Chr*?./02_out_byPostion/")
????system("rename?:?_?./02_out_byPostion/*")
????system("rename?-?_?./02_out_byPostion/*")
??}
??cli::cli_text("INFO???提取完成,對結果進行打包壓縮")
??system(str_c("tar?-czvf?",format(Sys.Date(),?"%Y_%m_%d"),"_",JOB,"_ExportFrom_",db_name,
???????????????"_AllSample_Filter_ByPositin",".tar.gz?./02_out_byPostion/*?./Tips.pdf"))
??cli::cli_text("INFO???任務運行結束,請及時下載結果文件,下次運行前將清空結果文件")
}
今天分享的內容就到這里,還有兩個功能正在開發中,之后有時間再分享關于提取指定位點名稱和結構注釋的方法,目前這項工具還未完成,如需搶先體驗請后臺聯系,后續將開源至Github,歡迎轉發分享。
參考資料:
https://mp.weixin.qq.com/s/DdXyqiW7c7lCp4103flQZQ
本文由 mdnice 多平臺發布




)


——CentOS安裝Kibana)



)

——現有網絡模型的使用及修改)



; 及 $grid->column(‘image‘);)
)
