題外話:
我踩過的坑,都將成為我寫貼的素材!(ㄒoㄒ)
整整安裝了兩天,這里面的滋味懂的都懂。
希望開發作者持續維護。
希望有人或者作者持續打包成sigularity鏡像使用,并且直接傳到github上,傳到docker上下載到linux上也好困難啊(ㄒoㄒ) ,這樣就不會吃conda安裝的苦了。
核心思想:
- 用mamba 或者conda 安裝,這樣依賴包都同時安裝好了。
- 網要好。我的實踐是早上網最好,比如7-9點時,我的下載網速最高能達到1.2M/s (因為就算設了清華源等國內源,還是有一部分需要鏈接到國外網址的,這個時候網速就十分受限。所以需要與國外上班族錯峰使用。)
安裝過程中出現這個報錯就是網不好造成的。👉conda或mamba install 相關軟件報錯 - 安裝data.table版本為1.14.8
安裝過程中報錯
Error: package or namespace load failed for 'methylKit':object 'key<-' is not exported by 'namespace:data.table'
詳細解釋看后續部分內容。
解決辦法就是安裝data.table到1.14.8或更低版本。👉2025.06.23【甲基化】|methylKit常見疑難解答與實用技巧(FAQ)
但是博主的方法我用了不成功,所以這里提供另一種方法。
正式開始:
- 創建環境并安裝所需包,部分細節看👉被迫在linux上用R(真的很難用啊)之如何在linux上正常使用R
mamba create -n methylkit_5 -c bioconda -c conda-forge bioconductor-methylkit bioconductor-genomation r-data.table=1.14.8 -y
bioconductor-methylkit 必須安裝
bioconductor-genomation 是為了后續將差異甲基化位點注釋到基因組上
r-data.table=1.14.8 必須指定版本安裝,讓mamba自動匹配合適的methylkit和genomation。(這就是我說的另一種方法,從安裝的時候就指定版本,這是我折騰最久的地方,不多說了。)
安裝關鍵過程:
+ bioconductor-methylkit 1.28.0 r43hf17093f_1 bioconda Cached
+ bioconductor-genomation 1.34.0 r43hf17093f_1 bioconda Cached
+ r-base 4.3.3 h65010dc_18 conda-forge Cached
+ r-data.table 1.14.8 r43h029312a_2 conda-forge Cache...
Downloading and Extracting Packages:Preparing transaction: done
Verifying transaction: done
Executing transaction: doneTo activate this environment, use$ mamba activate methylkit_5To deactivate an active environment, use$ mamba deactivate這里不禁要提一下原作者github上的issue部分:
原鏈接👉添加鏈接描述
防止有人打不開,這里再附一張截圖
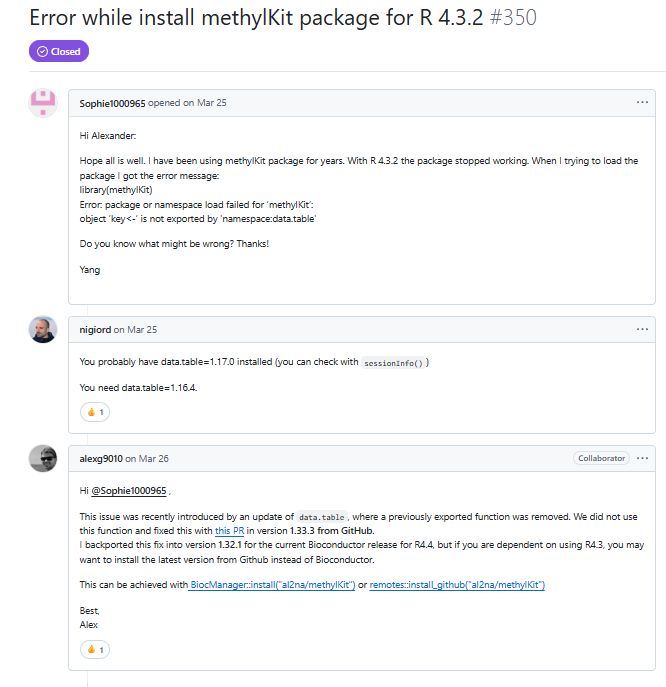
這里作者解釋了,這個報錯就是因為data.table更新了,新版本刪除了一個函數。作者修復了這一報錯,并建議從R版本為4.4的Bioconductor下載1.32.1版本的methylKit。(原文自己細看,這里是我的理解。)
但是bioconda里的版本截至到目前只更新到1.32.0(2025/8/29),這意味著linux上安裝methylkit建議還是安裝data.table的舊版本(1.14.8),methylkit安裝的版本也是舊版本,比如1.28.0。同時,也意味著,如果在電腦本地安裝,可以將R升級到4.4版本,然后Bioconductor就可以安裝methylkit的最新版本了。
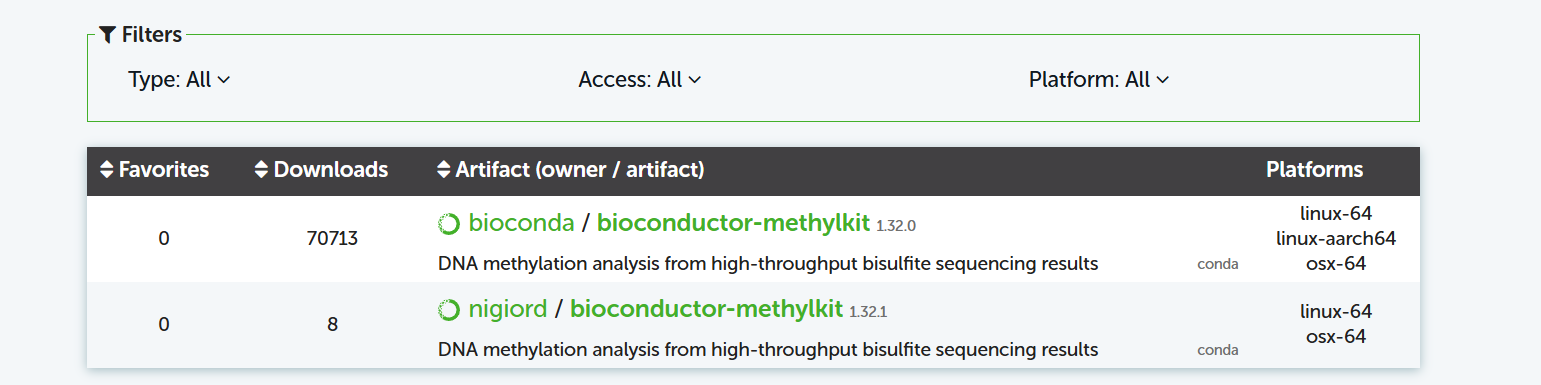
(nigiord的源可能不靠譜,不建議嘗試,頭鐵也可以試試安裝看下有沒有問題。)
- 檢測是否安裝成功
① 激活環境
mamba activate methylkit_5
② 打開R
直接命令行中輸入R,進入R
輸入.libPaths() #查看當前R包路徑
臨時改成該環境methylkit_5的R包路徑
.libPaths(c("/storage2/zuozhe/mambaforge/envs/methylkit_5/lib/R/library", .libPaths()))
這里每個人的路徑都不一樣,如果不確定可以去翻mambaforge/envs/methylkit_5/lib/R/library
就能得到自己的絕對路徑了。
③ library對應包,如果沒出現報錯說明就安裝成功了,正常使用。
(methylkit_5) zuozhe@server:~$ RR version 4.3.3 (2024-02-29) -- "Angel Food Cake"
Copyright (C) 2024 The R Foundation for Statistical Computing
Platform: x86_64-conda-linux-gnu (64-bit)R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> .libPaths()
> .libPaths(c("/storage2/zuozhe/mambaforge/envs/methylkit_5/lib/R/library", .libPaths()))
> library(methylKit)
Loading required package: GenomicRanges
Loading required package: stats4
Loading required package: BiocGenericsAttaching package: 'BiocGenerics'The following objects are masked from 'package:dplyr':combine, intersect, setdiff, unionThe following objects are masked from 'package:stats':IQR, mad, sd, var, xtabsThe following objects are masked from 'package:base':Filter, Find, Map, Position, Reduce, anyDuplicated, aperm, append,as.data.frame, basename, cbind, colnames, dirname, do.call,duplicated, eval, evalq, get, grep, grepl, intersect, is.unsorted,lapply, mapply, match, mget, order, paste, pmax, pmax.int, pmin,pmin.int, rank, rbind, rownames, sapply, setdiff, sort, table,tapply, union, unique, unsplit, which.max, which.minLoading required package: S4VectorsAttaching package: 'S4Vectors'The following objects are masked from 'package:dplyr':first, renameThe following object is masked from 'package:utils':findMatchesThe following objects are masked from 'package:base':I, expand.grid, unnameLoading required package: IRangesAttaching package: 'IRanges'The following objects are masked from 'package:dplyr':collapse, desc, sliceLoading required package: GenomeInfoDbAttaching package: 'methylKit'The following object is masked from 'package:dplyr':select> library(genomation)
Loading required package: gridAttaching package: 'genomation'The following objects are masked from 'package:methylKit':getFeatsWithTargetsStats, getFlanks, getMembers,getTargetAnnotationStats, plotTargetAnnotationWarning message:
replacing previous import 'Biostrings::pattern' by 'grid::pattern' when loading 'genomation'④ 如果library過程或者運行methylkit中報錯了缺啥包,就缺啥包再安裝啥包 (Warning message 影響不大)。這種情況沒什么更好的經驗,可以嘗試在R中直接安裝,也可以在bioconda中找到對應的包安裝,再重新library。如果折騰來折騰去都沒成功,建議重新建個環境,加上所需的包,比如我的就要求加上dplyr包,所以最終我的命令是:
mamba create -n methylkit_5 -c bioconda -c conda-forge bioconductor-methylkit bioconductor-genomation r-data.table=1.14.8 r-dplyr -y
目前就是這樣,研究methylkit命令去了。沒看到最后不知道還有個最終命令吧。











深入解析)



-轉載官方翻譯(有修正))



