作者簡介: 劉源 北京大學化學與分子工程學院/力文所
零.導讀
近幾年,蛋白質結構預測領域連續取得重大突破。首先是【AlphaFold】,在可以充分利用共進化信息結合深度神經網絡生成空間約束條件并降低相空間的搜索,極大地幫助了蛋白質的結構建模,顛覆了往年需要結合復雜結構采樣的算法,現在直接使用能量最小化即可得到預測的結構。隨后,在2019年底,David Baker團隊發表了【trRosetta】,其集合深度學習的諸多進展,并與Rosetta建模軟件結合,使得預測蛋白結構的門檻大大降低(在筆記本折疊蛋白) 。在【trRosetta】的文章中, 作者還發現了一個有趣的現象,對于很多之前設計的de novo design 的人工蛋白,在沒有同源序列(MSA)的情況下,只憑單序列輸入就可以預測到比較可靠的結構。
這個結果似乎暗示,trRosetta模型不但學到了用共進化信息來推斷空間約束,也學會了某些序列和結構之間的本質關聯。于是作者提出了兩個問題,
- 這些信息能否用來生成與訓練集序列不相關的新蛋白?
- 對于給定的空間約束(結構),模型能否通過反向傳播優化序列,也就是實現“design”的操作?
大佬David Baker 和 Sergey Ovchinnikov (Rising Star)最近在bioRxiv一起上線了兩篇文章,肯定了這兩個回答。在這兩篇文章中,同時也發現了一些令人意外的現象,讓我們一起來看看吧。
一、引理
蛋白質的結構和序列之間的關系,可以用條件概率和貝葉斯公式表示
P(seq|struct) = P(seq,struct)/P(struct) = P(struct|seq)*P(seq)/P(struct)
其中P(struct|seq),給定序列求結構,是trRosetta解決的結構預測問題。P(seq)是序列與結構無關的概率,也就是天然蛋白中氨基酸的頻率。P(struct)是與蛋白序列無關的結構信息,即背景。在文章中,作者對背景噪音單獨訓練了一個神經網絡,神經網絡的結構和trRosetta相似,但輸入的MSA為只與蛋白長度相關的隨機噪音。
二、不給結構隨便幻想
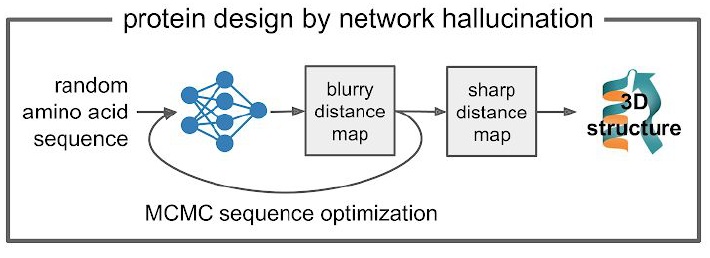
有了這個簡單的概率模型,作者的第一個問題就是如何讓神經網絡去幻想(hallucination)新的蛋白結構,我們能否隨機在P(seq,struct)中找一個位置然后找到它附近的極值?這就要說到深度學習中的一個有趣的問題DeepDream(深夢)。
DeepDream
這個方法反映的是一個神經網絡是怎么“認識”世界的,當你訓練好一個圖像分類器后,輸入一張圖片,deepdream就在圖中拼命尋找符合它所認識物體的pattern并加以放大,最后得到一張非常魔幻的照片。

這也正是幻想蛋白希望得到的效果,如果給定一個條序列(比如一條隨機序列)時, trRosetta預測出來的空間約束往往是缺乏特征的。如果能夠像deepdream一樣在里面尋找像是理想蛋白的特征并加以強化,最后我們就能得到一個具有良好蛋白性質的空間約束及其序列。
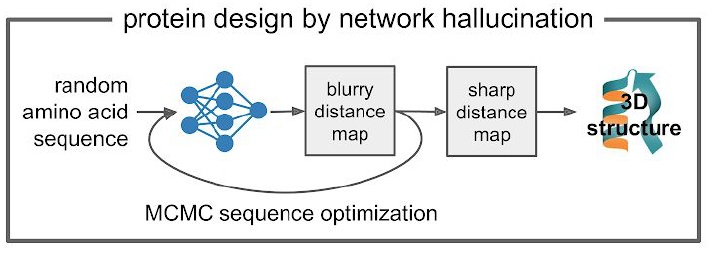
MCMC序列優化
具體的做法也相當簡單,首先給定一條初始序列(可以完全隨機也可以是有意義的序列),接著將一個大小為Lx64的隨機噪音輸入背景網絡,得到背景的空間約束。然后從初始序列出發,通過trRosetta網絡預測其空間約束,最初的約束可能分布相當彌散(因為序列不具有明顯結構特征),計算這個分布與背景分布的差異,如果兩者的KL散度越大,則說明得到的空間約束越像一個蛋白。作者在序列中隨機引入點突變,用Metroplis判據來不斷優化(增大)KL散度。
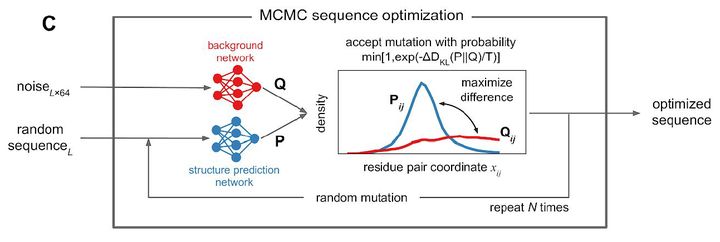
通過一個模擬退火的過程,隨著溫度不斷降低,作者得到了與噪音相比,KL散度非常大的序列。從D圖中可以看到,天然蛋白,從頭設計的蛋白,以及模擬退火后的幻象蛋白序列與背景空間約束分布的散度依次提高。40000步后幻想序列具有非常高的KL散度。
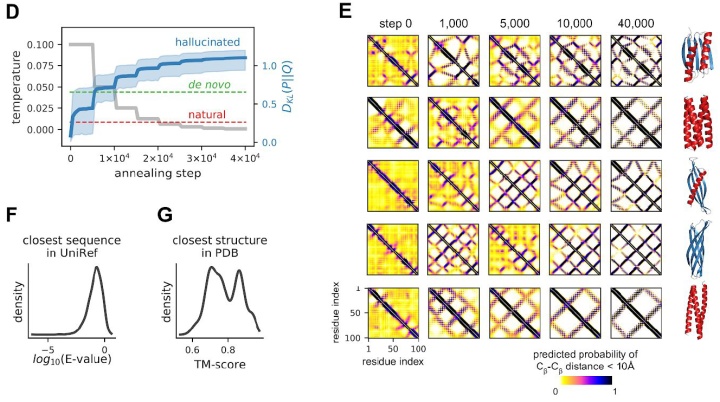
再比較一下序列,會發現幻想出來的序列距離天然蛋白非常遠,但結構上卻在PDB中具有很好的匹配程度。也就是說,幻想出來的蛋白是序列獨特但結構老套的蛋白。當然這并不奇怪,PDB庫早已經被報道覆蓋了蛋白質大部分可能折疊的空間,更何況trRosetta就是基于PDB結構進行的訓練。最終作者展示了多種全新幻想出來的序列,形成覆蓋全α全β或兩者混合的各類拓撲結構。
三、給定結構幻想序列
蛋白質設計的目的則在于優化P(seq|struct)。而在第二章節的MCMC采樣過程中,每次隨機突變一個氨基酸的方法效率較低,并沒有用到深度學習的關鍵技術“反向傳播”。這個方法可以使我們根據目標分布與預測分布的差異,有目的地批量更新氨基酸,為了用連續變量編碼氨基酸,文中采用PSSM來描述一條序列。這套方法被稱為trDesign。
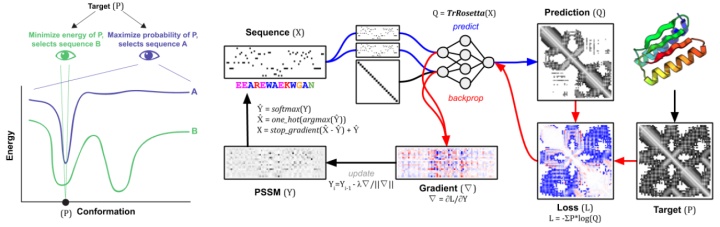
當我們有了一個目標結構的時候,就可以計算目標的空間約束(Target-P),用一條隨機序列通過trRosetta可以得到預測空間約束Q,P和Q的散度,就是我們希望最小化(使得Q接近P)的損失函數(注意在幻想時是希望最大化和背景的差異)。
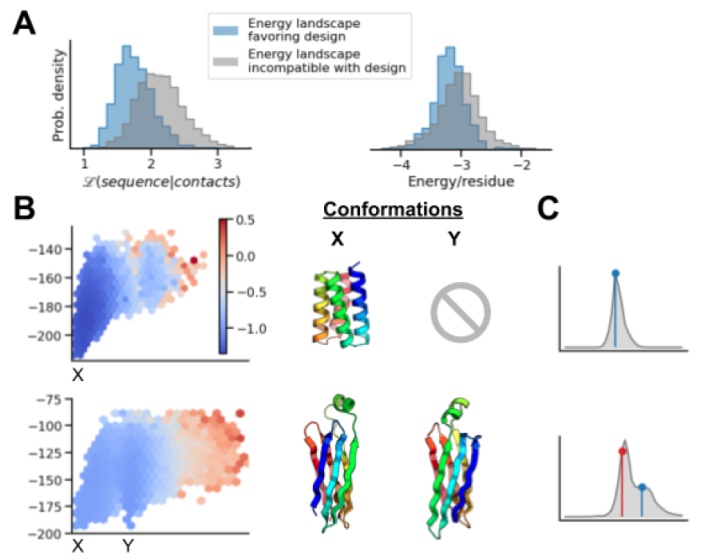
這個流程可以快速的對輸入序列進行優化,只需幾十步的迭代就可以得到收斂的結果。通過對Foldit玩家設計的幾千個蛋白進行分析,傳統的勢能面打分Pnear可以得到與實驗較好的關聯性,但開銷十分巨大。而trDesign的損失函數與Pnear有很好的關聯性,且對實驗驗證成功的例子有更好的區分度。說明trDesign所優化的是整個能量面,即降低目標構象的能量同時提高其它構象的能量。但缺點在于對native結構的優化不如Rosetta深入,這主要是受限于模型的精度。
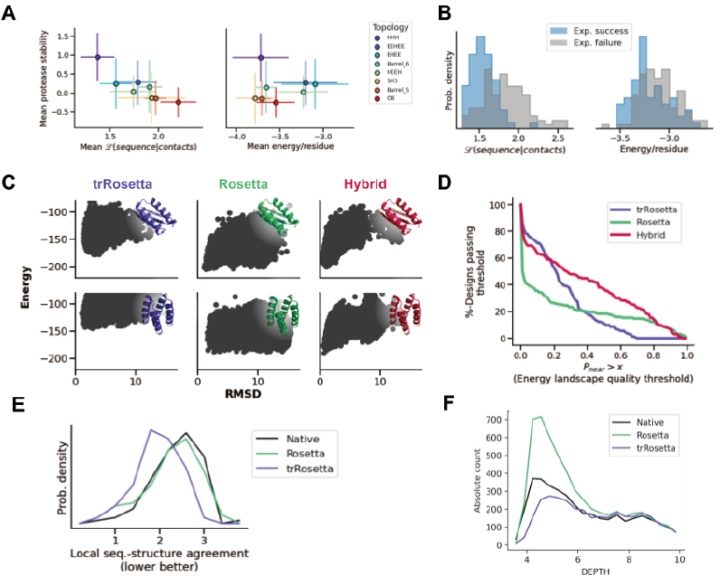
那么究竟trRosetta學到了什么呢?作者列出了三點:
- 一些距離的雙峰分布說明模型學到了全局或二級結構的不同堆疊狀態;
- 相對天然蛋白或De novo設計的蛋白而言,trRosetta設計的蛋白具有更理想的局域序列-結構關系(圖E);
- trDesign設計的蛋白具有更少的表面輸水側鏈(圖F),盡管它們對全局最小影響可能不大,但如果表面過多的疏水側鏈,這可能使得蛋白質會偏向折疊成使這些氨基酸包埋的亞穩態結構。
總結
作者開發的這套方法不但可以進行快速的蛋白質設計,而且其效果是可以優化整個能量面的形狀,如果與適合在局部深挖的Rosetta FastDesign相結合,可以達到遠超原來蛋白設計流程的效果。
隨心所欲
劉源:有人可能會說,你能想象出新蛋白又有什么用?能反向傳播又如何,不就比design快點么?于是重點來了,由于這兩個方法都是基于trRosetta模型來的,所以如果合理的設計損失函數,人們就可以做到固定一部分想要的結構,然后幻想生成剩下的部分!這也就是深度學習里常見的inpaint問題,擋住一部分圖片,自動補全新圖。而且最終得到的設計是非常接近ideal的穩定蛋白,具有很多好的性質,從這個角度trDesign可以看成是一種濾鏡,給輸入的蛋白“磨皮”,讓它更加完美(穩定易折疊,優化能量面)。最近的一個例子是Baker組設計的一個IL-2的mimic從頭設計蛋白【neo-2】,在文章中作者使用了大量復雜的算法生成主鏈構象再進行設計,而理論上這個操作可以在新的框架中一步到位。相信這套方案在成熟之后會在設計抗體、疫苗等重大問題上帶來突破性進展。
皮卡車:這兩篇文章的idea是如何出來的,和Sergey的一些經歷和想法分不開。Sergey 主要做的是共進化相關的工作,GREMLIN為主,也可以叫markov random field, potts model,self-supervised learning等。通過對MSA的分析來得到蛋白質的接觸圖譜。然后遇到的一個問題是,目前的結果是通過分析單層神經網絡的參數獲得的。如果層數增加,物理意義不明晰,就在模型中丟失了接觸圖譜的解釋性,于是他搞了一套基于梯度的分析方法,Seqsal,把輸入當變量,就可以從多層神經網絡中,得到蛋白質的接觸圖譜,于是各種模型,autoencoder,VAE等都可以通過這個方法來重新解析。在后來,trRosetta有了,是一種從序列到結構的分析方法。那么倒過來把序列當變量,通過調整序列來降低模型的損失函數,同時又把序列推離序列噪音,deep network hallucination就出來了。在損失函數中增加一項給定的結構約束,trDesign就出來了。在有了這些想法之后,能夠在幾個月時間內快速推進算法和實驗。除了baker實驗室,其他地方也難找了。
嘗鮮
現在已經提供了源代碼和例子,感興趣的同志可以去嘗試 https://github.com/gjoni/trDesign。 安裝和使用都比較簡單,但目前的例子也比較簡單,更復雜的功能有待諸君開發。

引文
- Deep network hallucination:I Anishchenko, TM Chidyausiku, S Ovchinnikov, SJ Pellock, D Baker. De novo protein design by deep network hallucination. (2020) bioRxiv, doi:10.1101/2020.07.22.211482
- trDesign:C Norn, B Wicky, D Juergens, S Liu, D Kim, B Koepnick, I Anishchenko, Foldit Players, D Baker, S Ovchinnikov. Protein sequence design by explicit energy landscape optimization. (2020) bioRxiv, doi:10.1101/2020.07.23.218917
- AlphaFold:A.W., Evans, R., Jumper, J. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020).
- trRosetta: Jianyi Yang, Ivan Anishchenko, Hahnbeom Park, Zhenling Peng, Sergey Ovchinnikov, and David Baker PNAS January 21, 2020 117 (3) 1496-1503
- neo-2:Silva, D., Yu, S., Ulge, U.Y. et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature 565, 186–191 (2019). https://doi.org/10.1038/s41586-018-0830-7





...)













