ml dl el學習
Application of Machine Learning and Deep Learning for Drug Discovery, Genomics, Microsocopy and Quantum Chemistry can create radical impact and holds the potential to significantly accelerate the process of medical research and vaccine development, which is a necessity for any pandemic like Covid19.
機器學習和深度學習在藥物發現,基因組學,顯微技術和量子化學中的應用可以產生根本性的影響,并具有顯著加速醫學研究和疫苗開發過程的潛力,這對于像Covid19這樣的大流行都是必不可少的。
Before we even begin, this article is a very high level one and specifically targeted for Data Scientists and ML Researchers interested in Drug Discovery, especially during the time of an existing pandemic like Covid19. If you are some one with a strong background in Bio-informatics or Chem-informatics and wants to venture into the world of data science for these use-cases, please reach out to me through any of the options mentioned here, and we can discuss few interesting opportunities for the greater good of mankind.
在我們甚至還未開始之前,本文是一篇非常高的文章,專門針對對藥物發現感興趣的數據科學家和ML研究人員,特別是在Covid19等現有大流行期間。 如果您是具有生物信息學或化學信息學背景的人,并且想涉足這些用例的數據科學領域,請通過此處提及的任何一種方法與我聯系,我們可以討論為人類帶來更大利益的機會很少。
DeepChem, an open source framework, which internally uses TensorFlow, that has been specifically designed to simplify the creation of deep learning models for various life science applications.
DeepChem是一個開放源代碼框架,內部使用TensorFlow,該框架專門設計用于簡化各種生命科學應用程序的深度學習模型的創建。
In this tutorial, we will see how to setup DeepChem and we will see how to use DeepChem for :
在本教程中,我們將了解如何設置DeepChem,以及如何將DeepChem用于:
1. training a model that can predict toxicity of molecules
1.訓練可以預測分子毒性的模型
2. training a model to predict solubility of molecules
2.訓練模型以預測分子的溶解度
3. using SMART strings to query molecular structures.
3.使用SMART字符串查詢分子結構。
設置DeepChem (Setting Up DeepChem)
Although, in multiple sources, I have seen users have expressed their concern in setting up DeepChem in Windows, Linux and Mac environments, but I found it quite easy to do that using pip installer.
雖然我從多個方面看到用戶已經表達了他們對在Windows,Linux和Mac環境中設置DeepChem的擔憂,但是我發現使用pip安裝程序非常容易。
The DeepChem development team, is very active and they do provide daily builds, so I would like everyone to take a look at their pypi page: https://pypi.org/project/deepchem/#history and install a suitable version in case if the latest version has any issue. A simple pip install deepchem would install the very latest version.
DeepChem開發團隊非常活躍,他們確實提供每日構建,因此,我希望每個人都可以查看他們的pypi頁面: https ://pypi.org/project/deepchem/#history并安裝合適的版本以防萬一如果最新版本有任何問題。 一個簡單的pip安裝deepchem將安裝最新版本。
Next, along with DeepChem, you would require TensorFlow to be installed. I had installed the latest version of TensorFlow using pip install tensorflow and RDkit which is an open source Cheminformatics software package. For RDkit and for installing in Windows, I did not find any reliable pip installer, so that installed it from https://anaconda.org/rdkit/rdkit using the conda installer : conda install -c rdkit rdkit
接下來,您需要與DeepChem一起安裝TensorFlow。 我已經使用pip install tensorflow和RDkit安裝了最新版本的TensorFlow,RDkit是開源的Cheminformatics軟件包。 對于RDkit和在Windows中安裝,我找不到任何可靠的pip安裝程序,因此使用conda安裝程序從https://anaconda.org/rdkit/rdkit安裝了它: conda install -c rdkit rdkit
Once these three modules are installed, we are ready to start with our experiments.
一旦安裝了這三個模塊,我們就可以開始實驗了。
預測分子的毒性 (Predicting toxicity of molecules)
Molecular toxicity can be defined as sum of adverse effects exhibited by a substance on any organism. Computational methods can actually determine the toxicity of a given compound using chemical and strcutral properties of the molecule and molecular featurization using molecular descriptors (Dong et al., 2015) and fingerprints (Xue and Bajorath, 2000), can effectively extract the chemical and structural information inherent in any given molecule for prediction-based approaches.
分子毒性可以定義為某種物質對任何生物產生的不利影響之和。 計算方法實際上可以使用分子的化學和結構特性確定給定化合物的毒性,并使用分子描述符(Dong等, 2015 )和指紋圖譜(Xue and Bajorath, 2000 )進行分子特征化,可以有效地提取化學和結構任何基于預測方法的給定分子固有的信息。
For predicting toxicity, we will use the Tox21 toxicity dataset from MoleculeNet and we will use DeepChem to load the required dataset.
為了預測毒性,我們將使用MoleculeNet的Tox21毒性數據集,并使用DeepChem加載所需的數據集。
import numpy as np
import deepchem as dc
tox21_tasks, tox21_datasets, transformers = dc.molnet.load_tox21()After this we will see all the toxicity classes, just be printing tox21_tasks
在此之后,我們將看到所有毒性類別,只需打印tox21_tasks
['NR-AR',
'NR-AR-LBD',
'NR-AhR',
'NR-Aromatase',
'NR-ER',
'NR-ER-LBD',
'NR-PPAR-gamma',
'SR-ARE',
'SR-ATAD5',
'SR-HSE',
'SR-MMP',
'SR-p53']We can divide the entire dataset into training, testing and validation dataset by:
我們可以通過以下方式將整個數據集分為訓練,測試和驗證數據集:
train_dataset, valid_dataset, test_dataset = tox21_datasetsIf we check the distribution of the dataset, we will see that the dataset is not balanced, so we would need to balance the dataset as typically we are trying to solve a MultiClass Classification problem. And so if the dataset is not balanced, the majority class will add bias to the classifier, which will skew the results. So, the transformer object used by default is a balancing transformer.
如果檢查數據集的分布,我們將看到數據集是不平衡的,因此我們將需要平衡數據集,因為通常情況下,我們正在嘗試解決“多類分類”問題。 因此,如果數據集不平衡,則多數類將為分類器增加偏差,這會使結果產生偏差。 因此,默認情況下使用的變壓器對象是平衡變壓器。
print(transformers)
[<deepchem.trans.transformers.BalancingTransformer at 0x26b5642dc88>]now, for the training part :
現在,對于培訓部分:
model = dc.models.MultitaskClassifier(n_tasks=12, n_features=1024, layer_sizes=[1000])
model.fit(train_dataset, nb_epoch=10)
metric = dc.metrics.Metric(dc.metrics.roc_auc_score, np.mean)
train_scores = model.evaluate(train_dataset, [metric], transformers)
test_scores = model.evaluate(test_dataset, [metric], transformers)Now, DeepChem’s submodule contains a variety of dc.models different life science–specific models.
現在,DeepChem的子模塊包含各種dc.models,不同生命科學特定的模型。
And finally we see, that the final AUC-ROC scores are:
最后我們看到,最終的AUC-ROC分數是:
{'training mean-roc_auc_score': 0.9556297601807405}
{'testing mean-roc_auc_score': 0.7802496964641786}This shows us that there is some over-fitting in the model as the test dataset metric scores are much less as compared to the train-set. But, nevertheless, now we do have a model that can predict toxicity from molecules!
這表明我們存在模型過度擬合的問題,因為與訓練集相比,測試數據集指標得分要低得多。 但是,盡管如此,現在我們有了一個可以預測分子毒性的模型!
預測分子的溶解度 (Predicting solubility of molecules)
Solubility is a measure, which shows how easily a molecule can dissolve in water. For any drug discovery, it is very important to check the solubility of the compound as the drug should dissolve into the patient’s bloodstream to have the required therapeutic effect. Usually, medicinal chemists spend a lot of time in modifying molecules to increase this property of solubility. In this section we will use DeepChem to predict solubility of molecules.
溶解度是一種度量,它顯示了分子在水中的溶解程度。 對于任何發現的藥物,檢查化合物的溶解度非常重要,因為藥物應溶解到患者的血流中以達到所需的治療效果。 通常,藥用化學家花費大量時間來修飾分子以增加溶解度的這種特性。 在本節中,我們將使用DeepChem預測分子的溶解度。
We will be using the delaney dataset from MoleculeNet, which is also available in DeepChem, for predicting molecular solubility.
我們將使用DeepChem中也提供的MoleculeNet的delaney數據集來預測分子溶解度。
# load the featurized data
tasks, datasets, transformers = dc.molnet.load_delaney(featurizer='GraphConv')# Split into traintest-validation dataset
train_dataset, valid_dataset, test_dataset = datasets# Fit the model
model = dc.models.GraphConvModel(n_tasks=1, mode='regression', dropout=0.2)
model.fit(train_dataset, nb_epoch=100)# Use r2 score as model evaluation metric
metric = dc.metrics.Metric(dc.metrics.pearson_r2_score)
print(model.evaluate(train_dataset, [metric], transformers))
print(model.evaluate(test_dataset, [metric], transformers))Even in the first pass, we see some overfitting from the model evaluation results.
即使在第一遍中,我們也會從模型評估結果中看到一些過擬合。
{'training pearson_r2_score': 0.9203419837932797}
{'testing pearson_r2_score': 0.7529095508565846}Let’s see how to predict solubility for a new set of molecules:
讓我們看看如何預測一組新分子的溶解度:
smiles = ['COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C',
'CCOC(=O)CC',
'CSc1nc(NC(C)C)nc(NC(C)C)n1',
'CC(C#C)N(C)C(=O)Nc1ccc(Cl)cc1',
'Cc1cc2ccccc2cc1C']Next, we need to featurize these new set of molecules from their SMILES format
接下來,我們需要從SMILES格式中使這些新的分子集特征化
from rdkit import Chem
mols = [Chem.MolFromSmiles(s) for s in smiles]
featurizer = dc.feat.ConvMolFeaturizer()
x = featurizer.featurize(mols)predicted_solubility = model.predict_on_batch(x)
predicted_solubilityAnd thus, we can see the predicted solubility values :
因此,我們可以看到預測的溶解度值:
array([[-0.45654652],
[ 1.5316172 ],
[ 0.19090167],
[ 0.44833142],
[-0.32875094]], dtype=float32)We saw very easily how DeepChem makes it very easy for the above two usecases, which may require alot of time for a human chemist to solve these problem!
我們非常輕松地了解到DeepChem如何輕松實現上述兩個用例,這可能需要很多時間才能讓化學工作者解決這些問題!
For the final part, we will see few visualizations and querying techniques available as a part of RDkit which is very much required when anyone is working for such use cases.
對于最后一部分,我們將看到很少的可視化和查詢技術可以作為RDkit的一部分使用,而在任何人使用此類用例的情況下,這都是非常必要的。
SMART字符串可查詢分子結構 (SMART strings to to query molecular structures)
SMARTS is an extension of the SMILES language described previously that can be used to create queries.
SMARTS是先前描述的SMILES語言的擴展,可用于創建查詢。
# To gain a visual understanding of compounds in our dataset, let's draw them using rdkit. We define a couple of helper functions to get startedimport tempfile
from rdkit import Chem
from rdkit.Chem import Draw
from itertools import islice
from IPython.display import Image, displaydef display_images(filenames):
"""Helper to pretty-print images."""
for file in filenames:
display(Image(file))def mols_to_pngs(mols, basename="test"):
"""Helper to write RDKit mols to png files."""
filenames = []
for i, mol in enumerate(mols):
filename = "%s%d.png" % (basename, i)
Draw.MolToFile(mol, filename)
filenames.append(filename)
return filenamesNow, let’s take a sample SMILES string and visualize the molecular structure.
現在,讓我們采樣一個SMILES字符串并可視化分子結構。
from rdkit import Chem
from rdkit.Chem.Draw import MolsToGridImage
smiles_list = ["CCCCC","CCOCC","CCNCC","CCSCC"]
mol_list = [Chem.MolFromSmiles(x) for x in smiles_list]
display_images(mols_to_pngs(mol_list))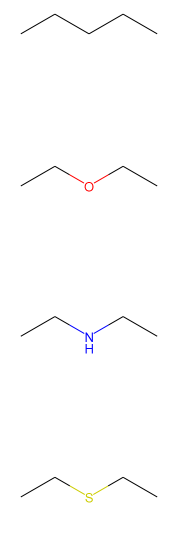
This is how the visual structures are formed from the SMILES string.
這就是從SMILES字符串形成視覺結構的方式。
Now, let’s say we want to query SMILES string that has three adjacent carbons.
現在,假設我們要查詢具有三個相鄰碳原子的SMILES字符串。
query = Chem.MolFromSmarts("CCC")
match_list = [mol.GetSubstructMatch(query) for mol in
mol_list]
MolsToGridImage(mols=mol_list, molsPerRow=4,
highlightAtomLists=match_list)
We see, that the highlighted part, represents the compound with three adjacent carbons.
我們看到,突出顯示的部分代表具有三個相鄰碳原子的化合物。
Similarly, let’s see some wild character query and other sub-structure query options.
同樣,讓我們??看一些通配符查詢和其他子結構查詢選項。
query = Chem.MolFromSmarts("C*C")
match_list = [mol.GetSubstructMatch(query) for mol in
mol_list]
MolsToGridImage(mols=mol_list, molsPerRow=4,
highlightAtomLists=match_list)
query = Chem.MolFromSmarts("C[C,N,O]C")
match_list = [mol.GetSubstructMatch(query) for mol in
mol_list]
MolsToGridImage(mols=mol_list, molsPerRow=4,
highlightAtomLists=match_list)
Thus, we can see that selective subquery can also be easily handled.
因此,我們可以看到選擇性子查詢也很容易處理。
Thus, this brings us to the end of this article. I know this article was very high level and specifically targeted for Data Scientists and ML Researchers interested in Drug Discovery, especially during the time of an existing pandemic like Covid19. Hope I was able to help! If you are someone with a strong background in Bio-informatics or Chem-informatics and wants to venture into the world of data science, please reach out to me through any of the options mentioned here. Keep following: https://medium.com/@adib0073 and my website: https://www.aditya-bhattacharya.net/ for more!
因此,這使我們到了本文的結尾。 我知道這篇文章的水平很高,特別針對對藥物發現感興趣的數據科學家和ML研究人員,特別是在Covid19等現有大流行期間。 希望我能提供幫助! 如果您是具有生物信息學或化學信息學背景的人,并且想涉足數據科學領域,請通過 此處 提到的任何一種方法與我聯系 。 繼續關注: https : //medium.com/@adib0073 和我的網站: https : //www.aditya-bhattacharya.net/ 了解更多 !
翻譯自: https://towardsdatascience.com/deepchem-a-framework-for-using-ml-and-dl-for-life-science-and-chemoinformatics-92cddd56a037
ml dl el學習
本文來自互聯網用戶投稿,該文觀點僅代表作者本人,不代表本站立場。本站僅提供信息存儲空間服務,不擁有所有權,不承擔相關法律責任。 如若轉載,請注明出處:http://www.pswp.cn/news/391182.shtml 繁體地址,請注明出處:http://hk.pswp.cn/news/391182.shtml 英文地址,請注明出處:http://en.pswp.cn/news/391182.shtml
如若內容造成侵權/違法違規/事實不符,請聯系多彩編程網進行投訴反饋email:809451989@qq.com,一經查實,立即刪除!相關文章

響應式網站設計_通過這個免費的四小時課程,掌握響應式網站設計

2017-2018-1 20179215《Linux內核原理與分析》第二周作業
實現)
python:單例模式--使用__new__(cls)實現
 自連接)
重學TCP協議(5) 自連接

Gradle復制文件/目錄方法

用戶參與度與活躍度的區別_用戶參與度突然下降
與__init__())
python:__new__()與__init__()
 四次揮手)
重學TCP協議(6) 四次揮手

mysql數據庫部分操作指令

推箱子2-向右推!_保持冷靜并砍箱子-嗶

UML建模圖實戰筆記

數據草擬:使您的團隊熱愛數據的研討會

python:列表推導式

WPF中刪除打開過的圖片
-文件系統)
深入理解InnoDB(5)-文件系統

vim捐贈_#PayItBackwards-一位freeCodeCamp畢業生如何向事業捐贈10,000美元

Digital River拉來Netconcepts站臺 亞太營銷服務升級


windows中怎么添加定時任務
)