完整內容請看文章最下面的推廣群

一、問題一:混合STR圖譜中貢獻者人數判定

- 問題解析
給定混合STR圖譜,識別其中的真實貢獻者人數是后續基因型分離與個體識別的前提。圖譜中每個位點最多應出現2n個峰(n為人數),但由于峰重合、共等位現象,實際峰數小于理論上限。 - 基礎模型
峰數-人數映射規則模型:
定義每個基因位點的觀測峰數為,理論人數估計可表達為:
其中為總基因位點數。該模型簡單快速,但不魯棒。
3. 高級模型:高斯混合模型(GMM)+AIC/BIC人數評估
模型假設
將每個位點的峰高(height)作為高斯混合變量,認為每位貢獻者在某些等位基因上形成峰高,整個圖譜服從若干高斯分布疊加。
建模流程
設定混合模型為:
對于不同的 n(假設貢獻者人數),用EM算法估計模型參數;
計算每個模型的 AIC/BIC 值:
選取最優 n 作為估計貢獻者數。
4. SCI常用方法
1.高斯混合模型(GMM) + BIC人數估計
?代表論文:
Perlin, M. W. (2009). “Explaining the likelihood ratio in DNA mixture interpretation.” Journal of Forensic Sciences.
利用混合峰高分布特征,通過最大似然估計構建 GMM 模型,結合 BIC/AIC 評估人數。
2.最大似然分解(MLD)
?用于反演最可能的混合人數,結合STR峰數與位點覆蓋情況。
?代表論文:
Cowell, R. G., Lauritzen, S. L., & Mortera, J. (2007). “A gamma model for DNA mixture analysis.” Bayesian Analysis.
3.變分貝葉斯推斷 (VB)
?比EM更穩定,處理高維混合峰建模。
?應用于DNA高維推斷,見于:
Journal of Computational Biology,Bioinformatics。
🔬推薦期刊/會議:
?Forensic Science International: Genetics
?Journal of Forensic Sciences
?IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB)
?Bioinformatics (Oxford Journal)
問題2 在分析出貢獻者人數后,還需要判斷各貢獻者的混合比例。當貢獻者比例接近時,等位基因可能重疊,導致誤判基因型。明確比例有助于更精準地分析混合圖譜。依據附件2中混合ST圖譜數據(如圖2所示)設計算法或模型,用于識別某一混合樣本中的貢獻者比例,并評估其準確性。
圖2 2人混合圖譜拆分示意圖
二、問題二:貢獻者比例識別模型
- 問題解析
比例識別涉及到從混合峰中解析各個體的DNA貢獻量,關鍵在于建模每個等位基因的峰高由多個個體的疊加形成。 - 基礎模型
線性系統擬合法:
設觀測峰高為,貢獻者比例為,每個貢獻者對峰高的貢獻可建模為:
其中為貢獻者i在等位基因j的表達強度(1或0)。
通過最小二乘擬合,解出。
3. 高級模型:非負矩陣分解(NMF)
STR混合峰數據構成一個 size × loci 的非負矩陣,我們使用NMF分解為:
其中:
:表示貢獻者比例;
:表示每位貢獻者在各等位基因上的影響。
NMF解法可通過乘法更新法或交替最小二乘法獲得。
4.SCI常用方法
1.非負矩陣分解(NMF)
將STR圖譜建模為非負組合,擬合比例與基因型成分。
代表論文:
Bleka, ?., Storvik, G. & Gill, P. (2016). “EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts.” Forensic Science International: Genetics.
2.貝葉斯分布比例建模(Bayesian Quantitative Contribution Estimation)
建立混合物比例的概率模型,計算各貢獻者對峰高的影響。
用于低比例個體建模,詳見:
Gill, P. et al. (2008). “DNA commission of the ISFG: recommendations on the interpretation of mixtures.” Forensic Sci. Int.: Genetics.
3.最大后驗估計(MAP)+ 馬爾科夫鏈蒙特卡洛(MCMC)
多用于構建比例的置信區間。
🔬推薦期刊/會議:
Forensic Science International: Genetics
Annals of Applied Statistics
Journal of the Royal Statistical Society
問題3 根據附件1與附件2的混合STR圖譜數據以及附件3中各個貢獻者的基因型,設計算法或模型,用于推斷某一混合STR圖譜中各個貢獻者對應的基因型,并評估其準確性。
三、問題三:基因型分離與個體識別
- 問題解析
目標是將混合樣本還原為若干基因型,并與已知數據庫中個體進行匹配。 - 基礎模型
基因型集合構造與最小距離匹配:
枚舉所有可能的基因型組合(若人數為n),定義樣本觀測峰與生成峰的最小歐氏距離作為評價指標,選取最小者作為估計組合。 - 高級模型:貝葉斯后驗匹配模型
模型設定
定義混合樣本為,候選基因型組合為,則后驗概率:
其中似然項為:
- 算法實現
利用 Gibbs Sampling 對候選基因型集合采樣;
對比各組合與附件3個體樣本基因型,匹配概率最高者作為識別結果。
5.SCI常用方法
1.貝葉斯個體識別框架(Bayesian Deconvolution)
?輸入混合圖譜,輸出最大后驗可能的個體組合。
?代表論文:
Cowell, R. G., Lauritzen, S. L., & Mortera, J. (2015). “Probabilistic expert systems for DNA mixture profiling.” Theoretical Population Biology.
2.Gibbs采樣 + 隱變量模型(Hidden Genotype Sampling)
?隱式考慮混合者的可能組合,每一代采樣更新后驗。
?代表模型系統:LikeLTD, EuroForMix。
3.深度圖神經網絡(GNN) + 序列標注結構
?建模基因型之間的依賴與條件結構,用于圖譜還原(新興研究)。
?相關應用初見于:
Bioinformatics,ISMB會議。
🔬推薦期刊/會議:
?Bioinformatics
?Forensic Sci Int: Genetics
?Journal of Computational Biology
?PLOS Computational Biology
問題4 依據附件4中混合STR圖譜數據(如圖3所示)設計算法或模型,用于減少混合樣本中噪聲的干擾,以提高混合樣本分析的準確性。
圖3 2人混合圖譜降噪示意圖
數據集及其說明見附件:
鏈接:https://pan.baidu.com/s/1aNpk0oONWA6w7JR7-PYGFg?pwd=3uu6 提取碼: 3uu6
四、問題四:去噪處理與圖譜清洗
- 問題解析
STR圖譜存在背景噪聲與偽峰,需提高信噪比以提升分析效果。 - 基礎模型
固定閾值法:
設定峰高閾值,低于閾值者視為噪聲。 - 高級模型:基于譜域變換+神經網絡濾波器
方法一:小波變換 + 閾值去噪
將峰高序列作小波變換;
對小波系數設定軟/硬閾值;
重構峰圖譜。
方法二:深度殘差卷積自編碼器(Denoising AutoEncoder, DAE)
輸入為原始峰圖譜;
輸出為預測無噪聲圖譜;
損失函數為MSE;
網絡結構采用ResNet殘差塊優化。 - SCI常用方法
1.小波變換+譜閾值濾波(Wavelet Denoising)
?通用于信號處理領域。對STR譜峰信號處理有顯著去噪效果。
?參考應用:
Chen, J. et al. (2017). “Application of wavelet transform for STR profile denoising.” Forensic Biology.
2.殘差自動編碼器(Denoising AutoEncoder, DAE)
?輸入原始譜,輸出重建譜,最小化MSE。訓練自監督。
?應用于本體建模相關工作:
Nature Methods,IEEE Transactions on Medical Imaging
3.變分自編碼器(VAE)+譜學習(Spectral Deep Models)
?VAE可建模峰值變動的潛變量,增強譜峰恢復。
?新興方向。
🔬推薦期刊/會議:
?Pattern Recognition Letters
?IEEE Transactions on Biomedical Engineering
?Nature Methods
?Bioinformatics
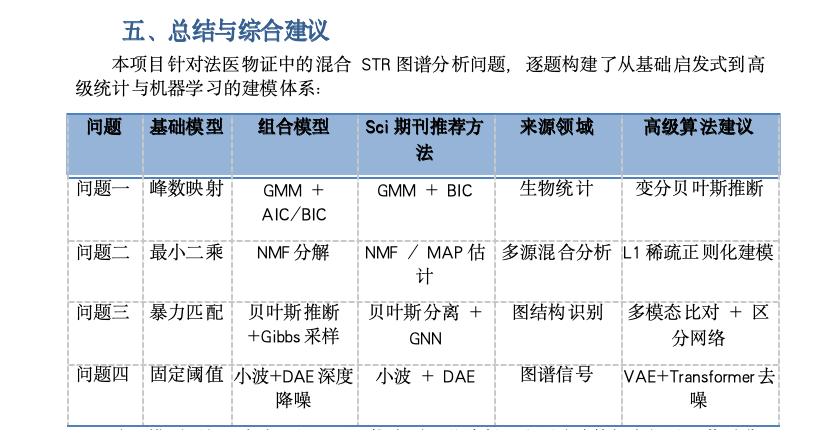
五、總結與綜合建議
本項目針對法醫物證中的混合STR圖譜分析問題,逐題構建了從基礎啟發式到高級統計與機器學習的建模體系:
問題 基礎模型 組合模型 Sci期刊推薦方法 來源領域 高級算法建議
問題一 峰數映射 GMM + AIC/BIC GMM + BIC 生物統計 變分貝葉斯推斷
問題二 最小二乘 NMF分解 NMF / MAP估計 多源混合分析 L1稀疏正則化建模
問題三 暴力匹配 貝葉斯推斷+Gibbs采樣 貝葉斯分離 + GNN 圖結構識別 多模態比對 + 區分網絡
問題四 固定閾值 小波+DAE深度降噪 小波 + DAE 圖譜信號 VAE+Transformer去噪
這些模型不僅具備實用性,還可推廣到醫學診斷、法證追蹤等復雜場景。若需進一步提升建模能力,可引入變分推斷、圖神經網絡(GNN)對等位基因關系建模,或生成對抗網絡(GAN)模擬生成圖譜增強訓練數據。


















在視頻模型中的幀間依賴建模)
