Pathview網站簡介
網址:https://pathview.uncc.edu/
前段時間介紹了一個R包 —?Pathview。它可以整合表達譜數據并可視化KEGG通路,操作是先自動下載KEGG官網上的通路圖,然后整合輸入數據對通路圖進行再次渲染。從而對KEGG通路圖進行一定程度的個性化處理,并且豐富展示信息。
Pathview包6大功能分別是:化合物和基因集同時繪制在通路上,多狀態或樣本同時或分開繪制,展示離散數據標記上下調或是否存在,不同來源的ID的轉換和映射,不同物種使用時名稱的處理和未注釋物種的處理 (直接用于宏基因組或微生物組數據)。具體見Pathview包:整合表達譜數據可視化KEGG通路
Pathview網站是在該包的基礎上,對一些核心功能進行延伸:
-
簡單直觀的圖形使用界面。
-
用的是
RESTful API,因此訪問速度很快。(該API是一個bash腳本,通過cURL使用。cURL是一個利用URL語法在命令行下工作的文件傳輸工具) -
有完整的通路分析流程,支持多組學數據和整合分析。
-
交互式并帶有超鏈接的結果圖能更好地解釋數據。
-
通過同步常規數據庫獲得最完整以及最新的通路數據。
-
所有資源和分析都是開源的。
-
注冊免費,登錄之后可以共享數據和保存分析歷史。
-
有完整的在線
Help和幫助文檔。 -
多個示例帶你快速上手使用。
輸入數據
輸入數據是最重要的且是唯一一個沒有默認值的選項。儲存數據矩陣的文件格式都是以tab或者逗號分隔的txt或者csv文件,點擊編輯框可設置對照組和處理組樣本。
數據類型主要分為兩類:
-
任何類型基因數據(表達譜、組蛋白修飾、染色質開放性等)的數據表,需要包含一列基因ID用于數據映射,比如
ENTREZ Gene,Symbol,RefSeq,GenBank Accession Number,Enzyme Accession Number等等,在選項框中共有13種基因ID可選。這里的基因數據是一個廣泛的概念,包括基因、轉錄本、蛋白質、酶及其表達、修飾和任何可測量的屬性。基因數據文件的第一列是基因ID,第一行是樣本ID。如果文件只有一列基因ID也是可以的。 -
化合物數據也是如此,包括代謝物、藥物、小分子和它們的測量值和屬性,以及用于數據映射的化合物ID,選項框中化合物數據庫ID共計22種,常用的是KEGG數據庫ID。除了行是化合物,化合物數據文件格式和基因數據文件的基本一致(或許還需要指定樣本列和實驗設計)。
輸出結果
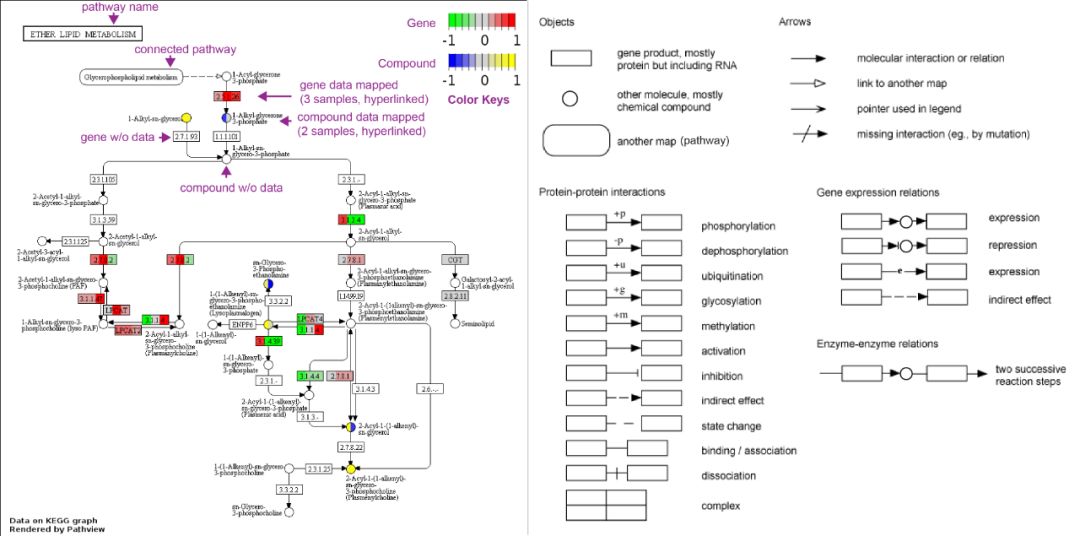
結果主要是數據整合得到的通路圖,有兩種:原始KEGG視圖和Graphviz視圖。
-
原始KEGG視圖將數據渲染到KEGG通路圖(柵圖,比如png格式),帶有大量的前后關系和元數據,解釋性更強。瀏覽器版本中該圖是可交互的,每個
Node都帶有超鏈接,可點擊它們轉到更詳細的解釋。
-
3種不同形狀表示不同的對象。
-
4種箭頭表示四種對象之間的關系。
-
12種蛋白質-蛋白質相互作用關系
-
4種基因表達之間的關系。
-
1種酶-酶關系:兩步連續反應。

-
Graphviz視圖是使用Graphviz引擎(矢量圖,如pdf格式)渲染的通路圖,在點/線屬性和圖形拓撲上更好理解。
-
16種連線類型
-
4種節點類型
操作
網頁版用示例展示了4個主要功能:多樣本的KEGG視圖,多樣本的Graphviz視圖,ID mapping和整合通路分析。最后一個會得到可視化文件和通路分析的結果統計文件(可下載),耗時較長。
該網站最突出的功能是ID mapping,整合的Mapper模塊將13種的基因或者蛋白質ID,22種化合物或者代謝物ID比對到標準KEGG的基因或化合物ID。換句話說,能將輸入的不同類型的數據ID精準比對到目標KEGG通路。
輸入和輸出選項/Input&Output
物種|Species:對應物種的KEGG號,科學名稱或公用名稱,比如可以在該選項中直接選擇KEGG Orthology的KO——ko-KEGG Orthology-N。常用的是hsa——home sapiens-human。具體根據導入的數據類型判斷。
通路選擇|Pathway Selection:對于連續型數據采用GAGE(Generally Applicable Gene-set Enrichment)方法或者離散型數據(比如基因或者化合物ID表)采用over-representation方法做通路分析(GO、GSEA富集分析一網打進)
GAGE是一種自限性原假設的基因集分析方法,充分利用了表達譜數據,并將表達數據分為實驗集和通路集分別進行分析處理,會考慮到基因集的上調和下調,得到更為準確和科學的結果。
如果沒有得到顯著的通路,會自動選擇靠前的幾個通路。基因數據和化合物數據一起分析的時候,會先各自篩選通路,然后通過meta分析將結果組合成更強大的全局統計量/ p值。
Pathway Selection一般建議選用auto,這在不確定通路的情況下再適用不過。若想自定義幾個通路,則可以選擇Manual。
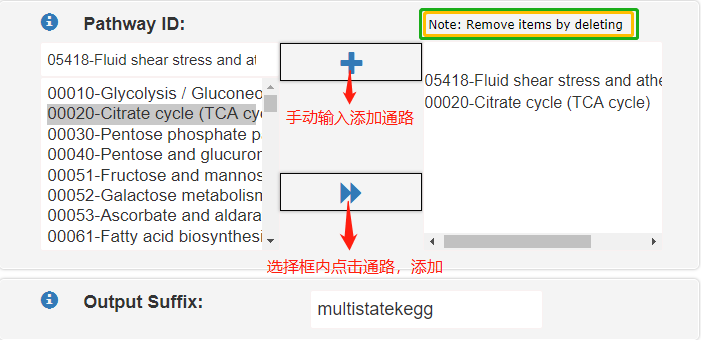
通路ID|PathwayID:是KEGG的通路ID,一般是5位數字,當通路選擇是auto時該選項自動關閉。
輸出后綴|Output Suffix:在結果文件名后面添加的后綴。
圖形選項/Graphics
Kegg Native:有KEGG圖形渲染(.png)和Graphviz引擎渲染(.pdf)。Graphviz引擎渲染可能會因為KEGG的xml數據文件缺失數據而丟失點。
Same Layer:圖層控制
-
Kegg Native項被勾選時,點的顏色會和通路圖在一個圖層,修改顏色的時候,節點標簽不變。
-
Kegg Native項未被勾選時,線/點類型的圖例會在一個圖層,節點標簽也會從原來的
KEGG基因標簽(或EC編號)變為官方基因符號。
離散型(基因和化合物數據)|Discrete:基因數據或者化合物數據一般是作為連續型數據使用。但也可以選擇被視為離散數據,這樣就可以以p值,倍數變化來選擇顯著的基因或者化合物列表,從而個性化標出離散數據中是否存在上下調。
但是網頁版本沒有設置選值的選項,還是Pathview包更適合使用這個設置。
Keys Alignment:當基因數據和化合物數據都不為NULL時如何對齊顏色標簽。默認選項為“ x”(由x坐標對齊)和“ y”(由y坐標對齊)。
多狀態|Multi State:默認值為TRUE,判定多狀態(指多個樣本或多列)基因數據或化合物數據是否應該整合并繪制在一張圖中。
換句話說,不勾選“Multi State”的情況下,基因或者化合物節點會切成多個來對應數據中的狀況數或者樣本數,即由”一張圖每個節點多種顏色”變為”多張圖每個節點一種顏色”。
數據匹配|Match Data:默認是TRUE,判定基因數據或化合物數據的樣本數是否匹配。
假設基因數據和化合物數據的樣本大小分別為m和n(m>n),多余的空列NA(不加顏色顯示)會在保證樣本大小一致的情況下添加部分到化合物數據中,如此,才能在Multi State為TRUE時,得到相同數量的基因節點和化合物節點片段。
Signature Position:pathview的署名位置,默認是左下角。選擇“None”的時候不顯示。
Key Position:顏色標簽的位置,默認是“左上角”。一般上面是基因節點,下面是化合物節點。選擇“None”的時候不顯示。
化合物節點名偏移|Compound Label Offset:設置化合物節點標簽在默認位置或者節點中心處的長度(僅在Kegg Native=FALSE時有用)。這個選項在化合物用全名標記時很實用,能決定化合物節點的外觀。
顏色選項/Coloration
節點計算|Node Sum:在比對有多基因或化合物時選擇計算節點總數的方法。默認值是Sum,還有mean,median,max,max.abs和random。
空值的顏色|NA Color:基因數據或者化合物數據中缺失值或NA值的顏色。選項有透明"transparent"和灰色?"grey"
限制(基因和化合物)|Limit (Gene and Compound):基因數據或化合物數據轉換為顏色時的限制值(即顏色標簽的數值范圍)。
這個選項是數值型的,一個框可以輸入用逗號分隔的兩個數字,比如“1,2”(不帶引號)—— 第一個數字表示下限,第二個數字表示上限。輸入單個值“n”的時候,網站認為范圍是(-n, n)。
Bins (Gene and Compound): 在基因數據和化合物數據轉換為顏色時,此參數可以設置顏色標簽的長度。預設值為10。
Low, Mid, High (Gene and Compound):低,中,高(基因和化合物),這些參數可以選擇“基因數據”和“化合物數據”的色譜。
“基因數據”和“化合物數據”的默認數據(低-中-高)分別是“綠色-灰色-紅色”和“藍色-灰色-黃色”。
這里既可以用顏色的通用名稱(綠色,紅色等),也可以用十六進制顏色代碼(比如00FF00,D3D3D3等)或顏色選擇器指定顏色。
網頁版優勢
以上是網頁版的參數選擇,較Pathview包而言少了Split Group|分組和擴展節點|Expand Node功能,個別參數的靈活性也待改進,但網頁版不需要占用本地內存,KEGG視圖的節點能超鏈接到更詳細的信息,而且多通路分析作為網頁版最大的優勢,有著完整的通路分析流程,支持多組學數據和連接公共通路。
這一步在本地的話還需要用gage包得到基因集在KEGG數據庫的所有通路分析結果,代碼如下:
> library(gage)
> data(gse16873)
> cn <- colnames(gse16873)
> hn <- grep('HN',cn, ignore.case =TRUE)
> dcis <- grep('DCIS',cn, ignore.case =TRUE)
> data(kegg.gs)
> #pathway analysis using gage 用gage做通路分析
> gse16873.kegg.p <- gage(gse16873, gsets = kegg.gs,
+ ref = hn, samp = dcis)
> #prepare the differential expression data 準備差異表達數據
> gse16873.d <- gagePrep(gse16873, ref = hn, samp = dcis)
> #equivalently, you can do simple subtraction for paired samples 得到成對的樣本
> gse16873.d <- gse16873[,dcis]-gse16873[,hn]
> #select significant pathways and extract their IDs 得到重要通路的ID
> sel <- gse16873.kegg.p$greater[, "q.val"] < 0.1 & !is.na(gse16873.kegg.p$greater[,
+ "q.val"])
> path.ids <- rownames(gse16873.kegg.p$greater)[sel]
> path.ids2 <- substr(path.ids[c(1, 2, 7)], 1, 8)
> #pathview visualization Pathview可視化
> pv.out.list <- sapply(path.ids2, function(pid) pathview(gene.data = gse16873.d[,
+ 1:2], pathway.id = pid, species = "hsa"))網頁版本實現方式如下——Example 4:

導入必要的數據,這里主要是設置了Pathway Selection為auto,選完之后點Submit就能得到完整分析結果,出來的頁面的中心處是設置說明,右側Completed下是分析結果和分析日志。
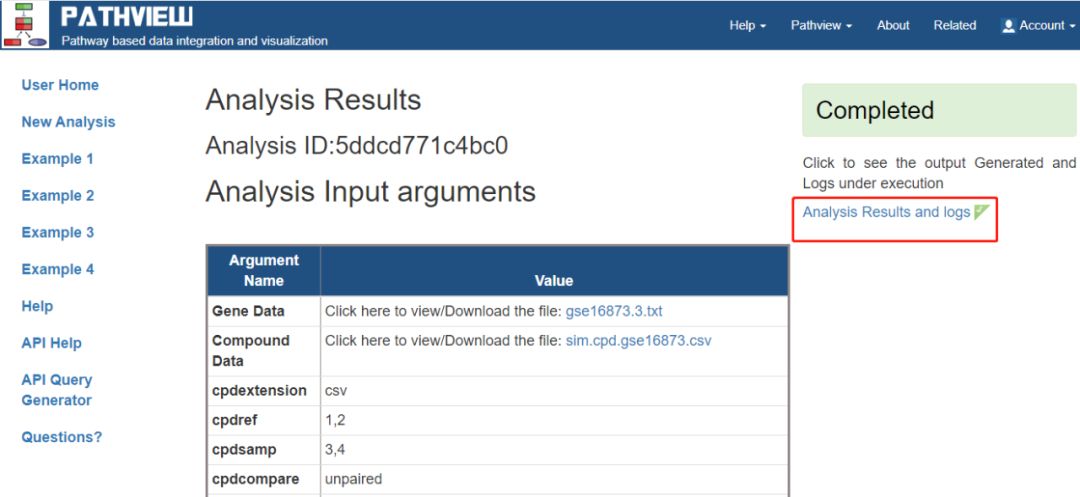
展示的是6種結果中的oxidative phosphorylation-氧化磷酸化代謝途徑的結果。

完整結果可看:
https://pathview.uncc.edu/resultview?analyses=5ddcd97621bd9&id=hsa-Homo%20sapiens&suffix=multistatekegg&autopathwayselection=True


)




安裝iptables防火墻)

——CRA角度)









