1. 基因分型Phasing概念
基因分型,也稱為基因定相、單倍體分型、單倍體構建等,即將一個二倍體(或多倍體)基因組上的等位基因(或雜合位點)正確定位到父親或母親的染色體上,最終使得來自同一親本的等位基因能排列在同一條染色體上。
二代測序是將序列混在一起進行測序,通過比對到參考基因組,檢測存在哪些變異以及變異的基因型(Genotype),但無法直接區分序列是母源還是父源,只有通過基因分型才能判斷親本來源。
2. 基因分型的方法
家系分型和LD分型常用的工具有Beagle和Shapeit,都包含了家系分型和LD分型模塊。
2.1 家系分型
家系分型是目前構建單倍型最準確的方法,家系樣本越多,分型效果越好,缺點就是需要家系Trio樣本,對于無法獲取家系樣本時則無法完成分型,并對于父親、母親和子女都是雜合的位點無法進行分型(大約占總變異位點的1/5)。
2.2 LD分型
利用群體中大量無血緣關系的個體,根據LD(連鎖不平衡)原理和數據模型,推斷群體中每個個體的單倍型方法。通過人群頻率大于5%的變異存在的LD block(Tajima’s D > 0.5)長度大多為50kbp-60kbp,block的長度在不同人種中不相同,非洲人由于更古老因此姐妹染色單體發生重組的次數會更多,LD block相比于其他人種長度更短。由于LD block的存在,因此可以利用數學模型(如HMM算法)反推出個體的單倍型。
LD分型精度受到群體的影響,對于人群頻率大于5%的變異分型效果很好,但對于罕見變異和低頻變異(<1%)的效果較差,因此很難獲得個體完整的單倍型。
2.3 物理分型
一條reads/一對reads或克隆上存在的堿基必定來自同一條染色體,每個片段就是單倍體的局部,將局部連接為整體即可完成Phasing。物理分型不需要家系數據,無需借助LD關系,僅依賴自身的測序數據,就可以完成Phasing。物理分型依賴于reads上的雜合SNP位點作為區分標記,由于人類基因組雜合SNP之間的距離大約為1.5kpb,因此需要長序列的測序技術(三代PacBio、ONT和華大Long Fragment Read-LFR等)獲取測序數據。
3. 遺傳解讀中的應用
在遺傳解讀時,需要獲取基因突變位點的相位后,才能更好地判斷突變是否會產生對應的表型。當一個等位基因的一個拷貝發生了變異(例如LOF,功能缺失或缺失等),由于存在另一個拷貝,基因表達可能不會受到影響,從而不具有表型,只有當兩個拷貝都發生變異時,才影響基因的表達從而產生表型。
4. 人類單倍型參考序列Reference Consortium(HRC)
基因型推斷(Imputation)
需要單倍型參考序列集作為基礎數據,在全基因組關聯分析GWAS中是必要的環節,參考基因序列的質量直接影響了后續基因型-表型關聯分析的結果。
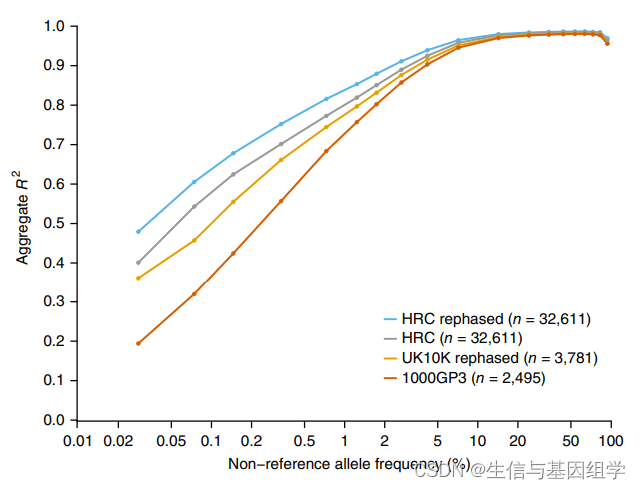
HRC是一個大型的人類單倍型參考序列,合并了多個項目的測序數據。全基因組SNP微陣列芯片獲得樣本基因型后,可使用該單倍型參考基因組進行基因型的推斷(或稱為填充)和定相,GWAS研究中運用較為常見。截止到2024年7月,HRC包含了64,976個人類單倍型和39,235,157個SNP。相比于1000G、UK10K數據庫,采用HRC單倍型數據庫基因型填充的準確率更高。
目前提供網頁版的免費的基因型推斷和定相服務-Sanger Imputation,網址如下:
https://www.sanger.ac.uk/tool/sanger-imputation-service/
基因型推斷的工具網站:
https://imputation.sanger.ac.uk/
sanger網站需要上傳VCF或23andMe格式的GWAS數據,預定相可選擇EAGLE2或SHAPEIT 2,基因型推斷PBWT算法,參考panels可選擇1000 Genomes Phase 3, UK10K和 the Haplotype Reference Consortium。
https://imputationserver.sph.umich.edu/
HRC 只公開了部分數據提供下載,通過Request Access下載,鏈接如下:
https://ega-archive.org/datasets/EGAD00001002729



















 L3 網格化 1 度年土壤濕度,第 5 版)
