許多大佬的軟件想要構建一個大而美的生態,從 monocle2 開始就能做單細胞的質控、降維、分群、注釋這一系列的分析,但不幸的是我們只知道 monocle 系列還是主要做擬時序分析,一方面是因為 Seurat 有先發優勢,出名要趁早,生態太過強大,另一方面 monocle2 和monocle3 軟件有自身的問題,很難安裝或者有很多 bug,遠沒有 Seurat 穩定。

什么是Monocle3?
不過怎么樣Monocle3 還是最常用的單細胞軌跡分析工具之一,它能夠通過算法學習細胞在動態生物學過程中基因表達變化的序列,從而構建出細胞狀態轉變的軌跡。與傳統實驗方法不同,Monocle3無需純化處于中間狀態的細胞,就能讓我們清晰地看到細胞從一種功能“狀態”向另一種狀態的過渡。
當細胞過程存在多種結果時,Monocle3會構建出“分支”軌跡,這些分支對應著細胞的“決策”過程。它還提供了強大的工具來識別受這些決策影響以及參與決策的基因,幫助我們深入理解細胞命運決定的分子機制。
Monocle3核心思想
無需通過實驗將細胞提純為離散狀態,而是借助算法學習細胞在動態生物學過程中必然經歷的基因表達變化序列,進而掌握基因表達變化的整體 “軌跡”,并確定每個細胞在該軌跡中的準確位置。
核心工作流程
Monocle3的軌跡重建工作流程與聚類分析流程相似,但增加了幾個關鍵步驟。下面我們詳細介紹其核心流程:
安裝
注意 linux、mac 安裝都需要編譯,那么就需要 gcc、gfortran等編譯軟件,確保已經安裝
sudo apt install build-essential pkg-config libgdal-dev -y
BiocManager::install(c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats','limma', 'lme4', 'S4Vectors', 'SingleCellExperiment','SummarizedExperiment', 'batchelor', 'HDF5Array','ggrastr'))
BiocManager::install("bnprks/BPCells/r")
BiocManager::install('cole-trapnell-lab/monocle3')
library(monocle3)
1. 數據準備與初始化
首先,需要將數據加載到Monocle3的cell_data_set(CDS)對象中,這是Monocle3處理數據的基本單位。CDS對象包含三個關鍵部分:表達矩陣、細胞元數據和基因注釋。
官網示例
library(monocle3)
# 加載數據
expression_matrix <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_expression.rds"))
cell_metadata <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_colData.rds"))
gene_annotation <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_rowData.rds"))# 創建CDS對象
cds <- new_cell_data_set(expression_matrix,cell_metadata = cell_metadata,gene_metadata = gene_annotation)
但是更多時候我們是從已經處理好的 Seurat 對象構建CDS對象
# srt是之前注釋好的Seurat 對象
cds <- monocle3::new_cell_data_set(expression_data = SeuratObject::GetAssayData(srt, assay = "RNA", layer = "counts"),cell_metadata = srt@meta.data,gene_metadata = data.frame(gene_short_name = rownames(srt), row.names = rownames(srt))
)
2. 數據預處理
預處理步驟與聚類分析完全相同,包括數據標準化和批次效應校正等。在批次校正中,可以使用align_cds()函數,并通過alignment_group指定批次分組
# 數據預處理
cds <- preprocess_cds(cds, num_dim = 50)
# 批次校正
cds <- align_cds(cds, alignment_group = "batch")
3. 降維與可視化
接下來進行數據降維,對于軌跡分析,強烈建議使用UMAP方法(默認方法)。降維后可以使用plot_cells()函數可視化結果,通過不同的顏色編碼展示細胞類型等信息。
# 降維
cds <- reduce_dimension(cds)用 Seurat 對象中的 UMAP 替換 CDS 中的
cds.embed <- cds@int_colData$reducedDims$UMAP
int.embed <- Embeddings(srt, reduction = "umap")
int.embed <- int.embed[rownames(cds.embed), ]
cds@int_colData$reducedDims$UMAP <- int.embed
可視化 UMAP 圖
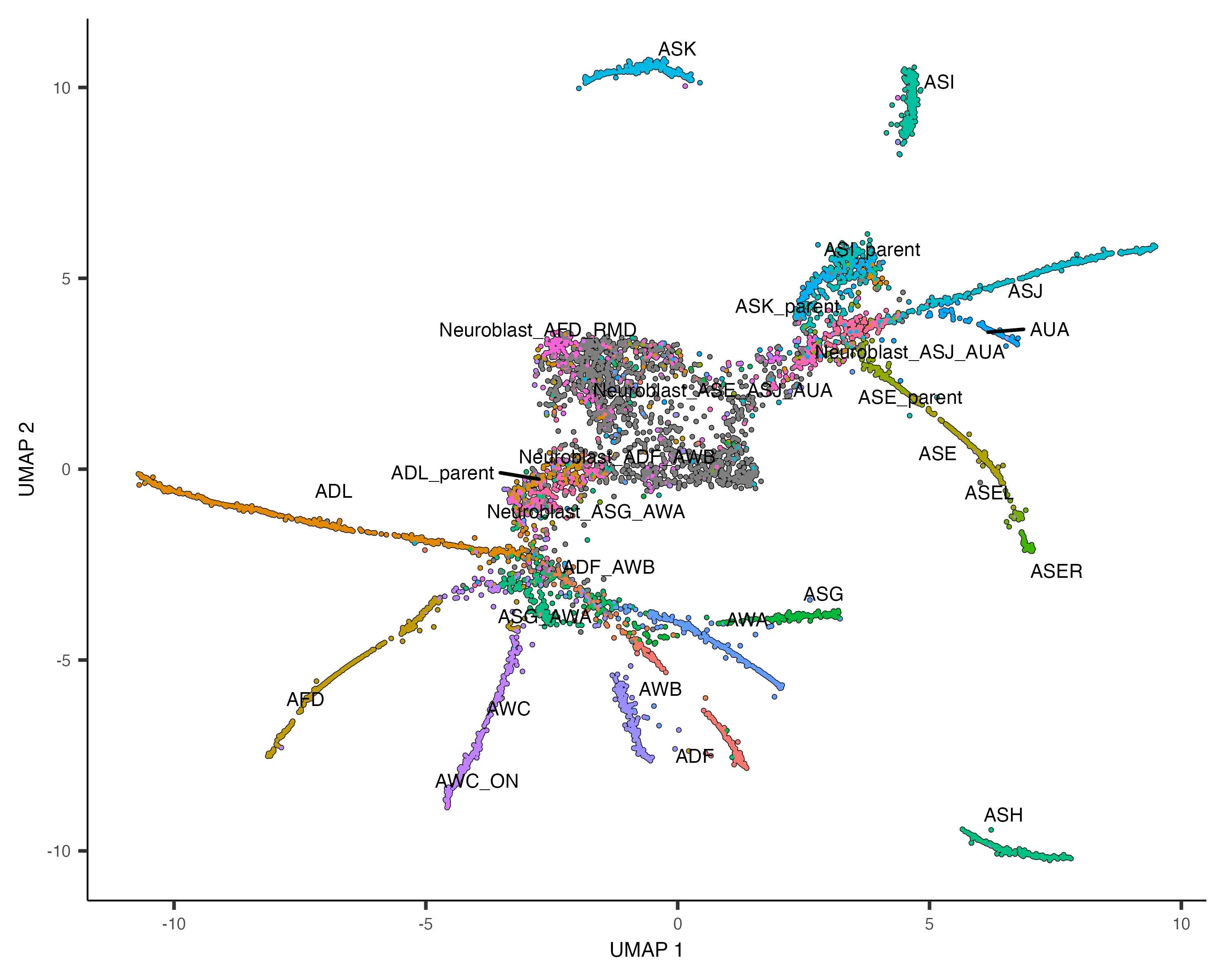
plot_cells(cds, label_groups_by_cluster = FALSE, color_cells_by = "cell.type")
通過這一步驟,我們可以初步觀察到數據中存在的軌跡結構,以及不同細胞類型在軌跡上的分布。
4. 細胞聚類
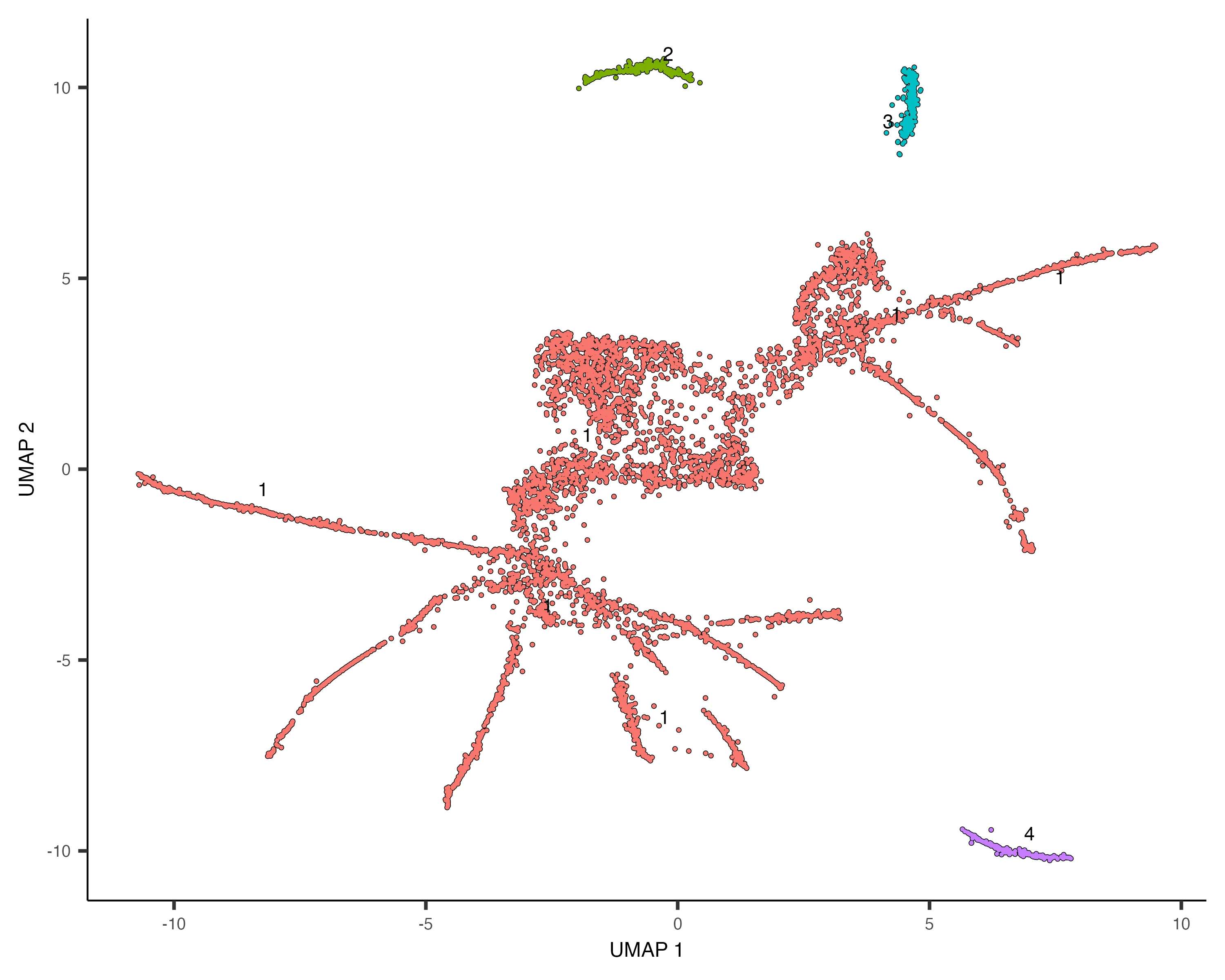
與聚類分析類似,使用cluster_cells()函數對細胞進行聚類。在軌跡分析中,每個細胞不僅被分配到一個聚類,還會被分配到一個分區(partition),每個分區最終會形成一個獨立的軌跡。
不同的分區可以有不同的分化起始點,這也符合現實情況,因為很可能這些細胞是多起源的。
# 細胞聚類
cds <- cluster_cells(cds)
# 按分區可視化
plot_cells(cds, color_cells_by = "partition")
5. 學習軌跡圖
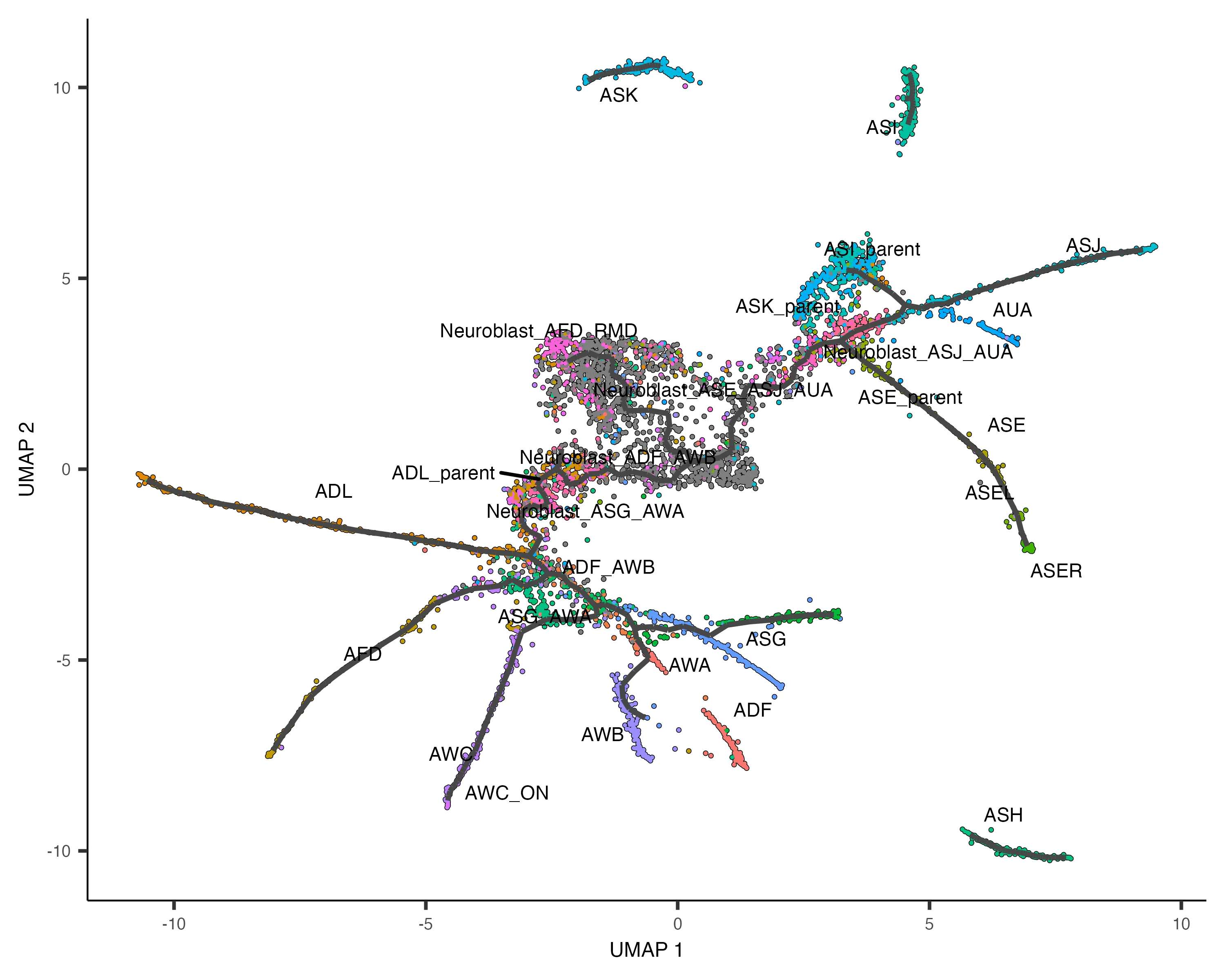
使用learn_graph()函數在每個分區內擬合主圖(principal graph),該圖將用于后續的分支分析和差異表達分析等下游步驟。
# 學習軌跡圖
cds <- learn_graph(cds)
# 可視化軌跡圖
plot_cells(cds, color_cells_by = "cell.type", label_groups_by_cluster = FALSE, label_leaves = FALSE, label_branch_points = FALSE)
軌跡圖的構建是Monocle3軌跡分析的核心步驟之一,它能夠捕捉細胞狀態之間的連續過渡關系。
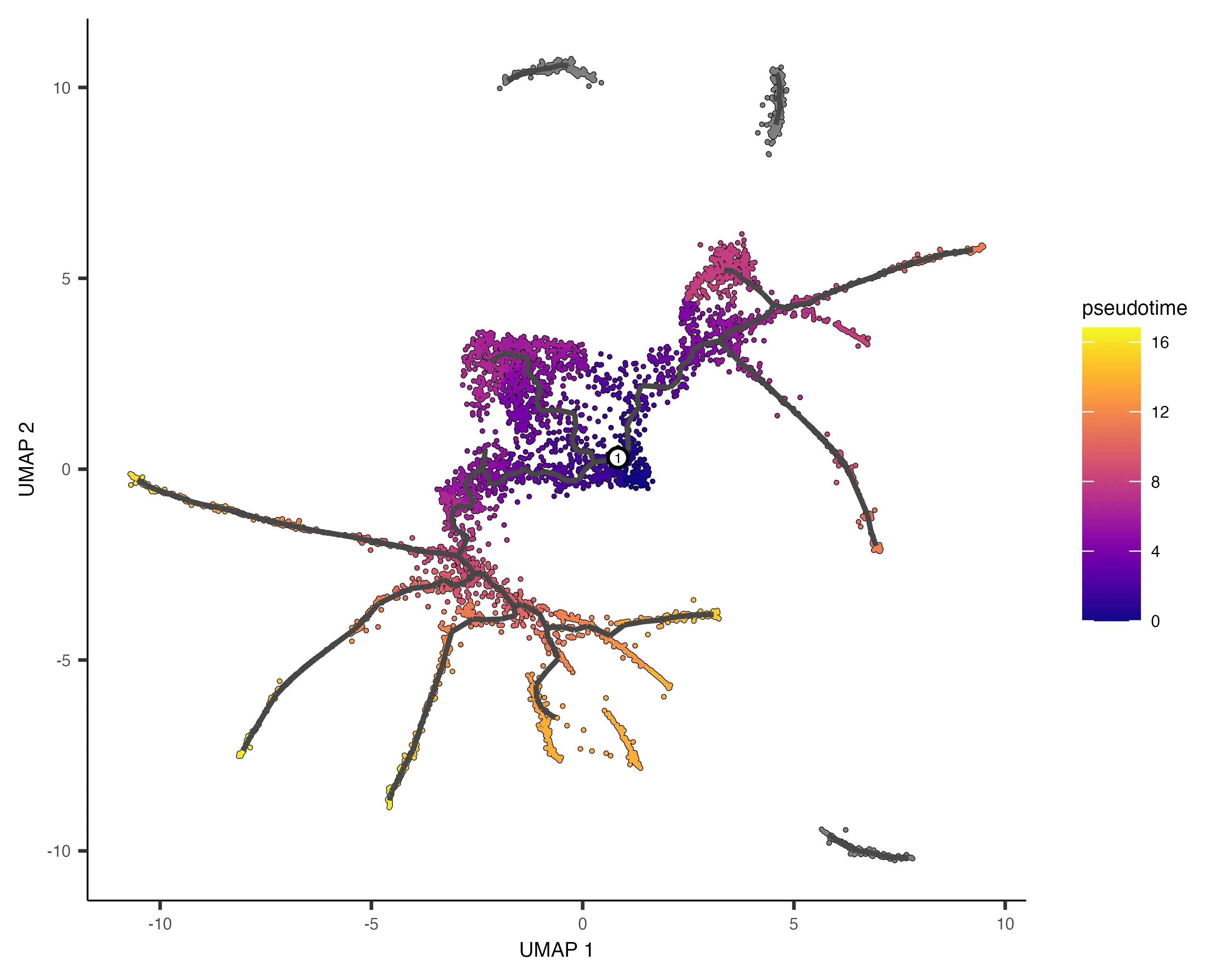
6. 以偽時間(Pseudotime)排序細胞
偽時間是衡量單個細胞在細胞分化等過程中進展程度的指標。通過order_cells()函數,我們可以根據細胞在軌跡上的進展對其進行排序。
- 什么是偽時間? 偽時間是一個抽象的進展單位,它表示細胞沿著學習到的軌跡從起始狀態到結束狀態所經歷的距離。在許多生物學過程中,細胞的發育并不同步,偽時間很好地解決了這種不同步發育帶來的分析難題。
# 按偽時間排序細胞
cds <- order_cells(cds)
# 按偽時間可視化
plot_cells(cds, color_cells_by = "pseudotime", ...)
在排序過程中,需要指定軌跡圖的根節點(root nodes),可以通過圖形界面手動選擇,也可以通過編程方式指定。例如,可根據最早時間點的細胞分布來自動確定根節點:
下邊這個函數可以幫助我們選擇發育起點,比如我們可以修改為從某一類細胞發育作為 root,只需要修改embryo.time.bin 為 cell.type 和time_bin 為我們想要設定的細胞類型
# 編程方式指定根節點
get_earliest_principal_node <- function(cds, time_bin="130-170"){cell_ids <- which(colData(cds)[, "embryo.time.bin"] == time_bin)closest_vertex <-cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertexclosest_vertex <- as.matrix(closest_vertex[colnames(cds), ])root_pr_nodes <-igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names(which.max(table(closest_vertex[cell_ids,]))))]root_pr_nodes
}
cds <- order_cells(cds, root_pr_nodes = get_earliest_principal_node(cds))
值得注意的是,未從根節點可達的細胞會被分配無限偽時間(顯示為灰色),因此通常應為每個分區至少選擇一個根節點。
7. 時序相關差異表達分析
Monocle3提供了強大的差異表達分析工具,可用于尋找隨著偽時間變化或在不同分支中表達發生變化的基因。例如:
可以選擇前 10 個基因展示,官網教程是自己選擇的與研究相關的基因
genes <- monocle3::graph_test(cds, neighbor_graph = "principal_graph", reduction_method = "UMAP", cores = 32)
top10 <- genes %>%top_n(n = 10, morans_I) %>%pull(gene_short_name) %>%as.character()
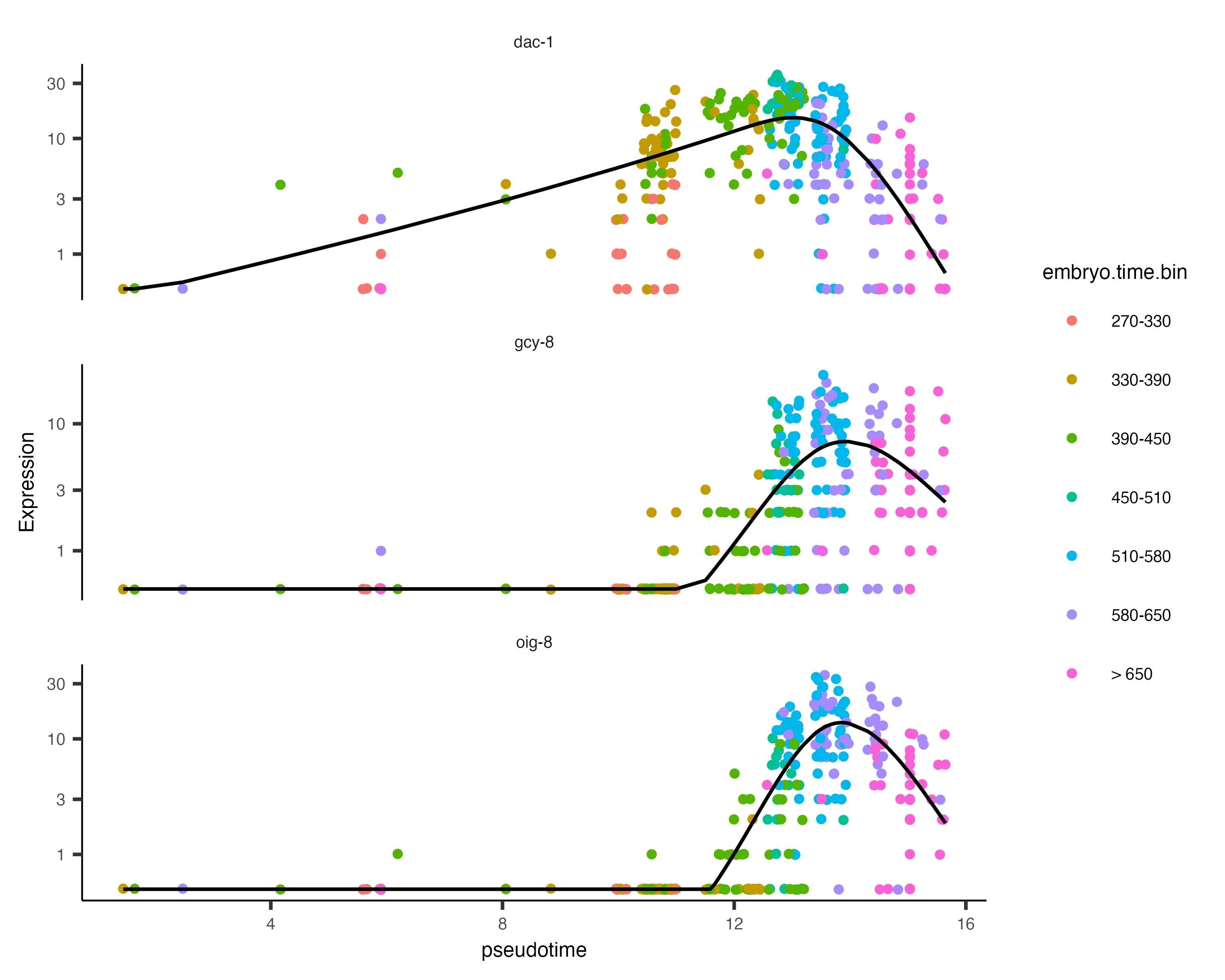
展示AFD_genes 基因隨時間變化圖
AFD_genes <- c("gcy-8", "dac-1", "oig-8")
AFD_lineage_cds <- cds[rowData(cds)$gene_short_name %in% AFD_genes,colData(cds)$cell.type %in% c("AFD")]
AFD_lineage_cds <- order_cells(AFD_lineage_cds)plot_genes_in_pseudotime(AFD_lineage_cds,color_cells_by="embryo.time.bin",min_expr=0.5)
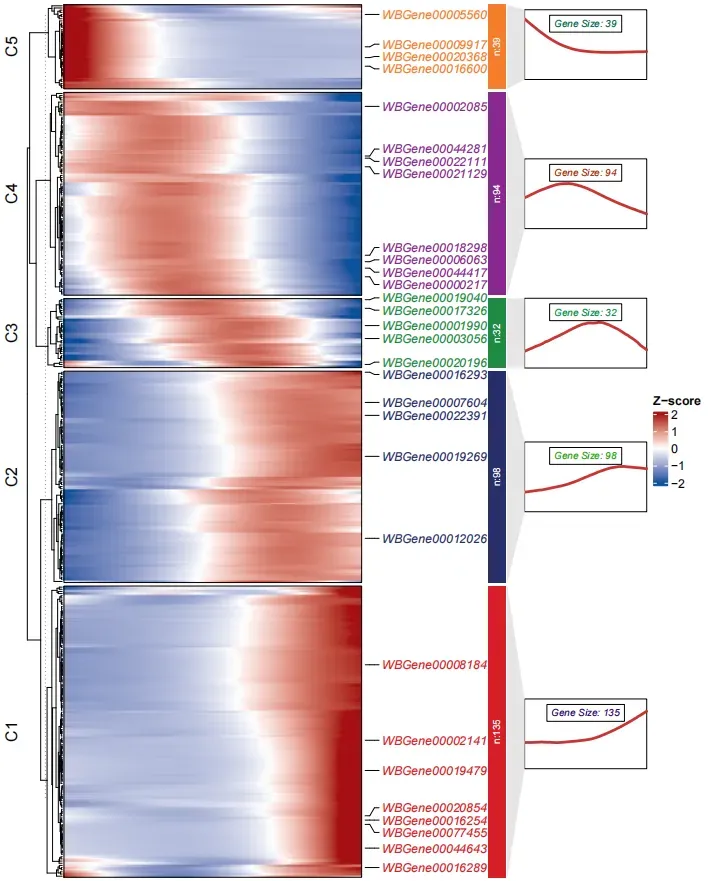
8. 擬時序基因熱圖
這里演示下選擇 50 個基因畫熱圖用 junjun 老師的 R 包ClusterGVis繪制,雖然 monocle3 有自己的分模塊熱圖,感覺沒啥用也不好看,不如這個更醒目一些。
top50 <- genes %>%top_n(n = 10, morans_I) %>%pull(gene_short_name) %>%as.character()
繪制熱圖,cluster.num 分 5 個 cluster 需要自己設置,markGenes 隨機選擇 30 個展示,可以自己設置標注哪些基因
library(ClusterGVis)# kmeans
ck <- clusterData(mat,cluster.method = "kmeans",cluster.num = 5)# add line annotation
pdf('monocle3.pdf',height = 10,width = 8,onefile = F)
visCluster(object = ck,plot.type = "both",add.sampleanno = F,markGenes = sample(rownames(mat),30,replace = F))
dev.off()
Reference
https://cole-trapnell-lab.github.io/monocle3/docs/differential/
https://mp.weixin.qq.com/s/7sYTrHlhnHj5490RyrdBug


介紹)











![[工具類] 網絡請求HttpUtils](http://pic.xiahunao.cn/[工具類] 網絡請求HttpUtils)
)
變換矩陣中的齊次坐標推導與幾何理解)


