From生物技能樹 GEO數據挖掘第一節
(pipeline)
文章目錄
- 1.圖表分析
- 2.GEO背景介紹及分析思路
- 3.代碼分析流程
- 4.復雜數據分析
- 理論知識
- 1.數據從哪里來
- 2.有什么類型的數據可挖掘
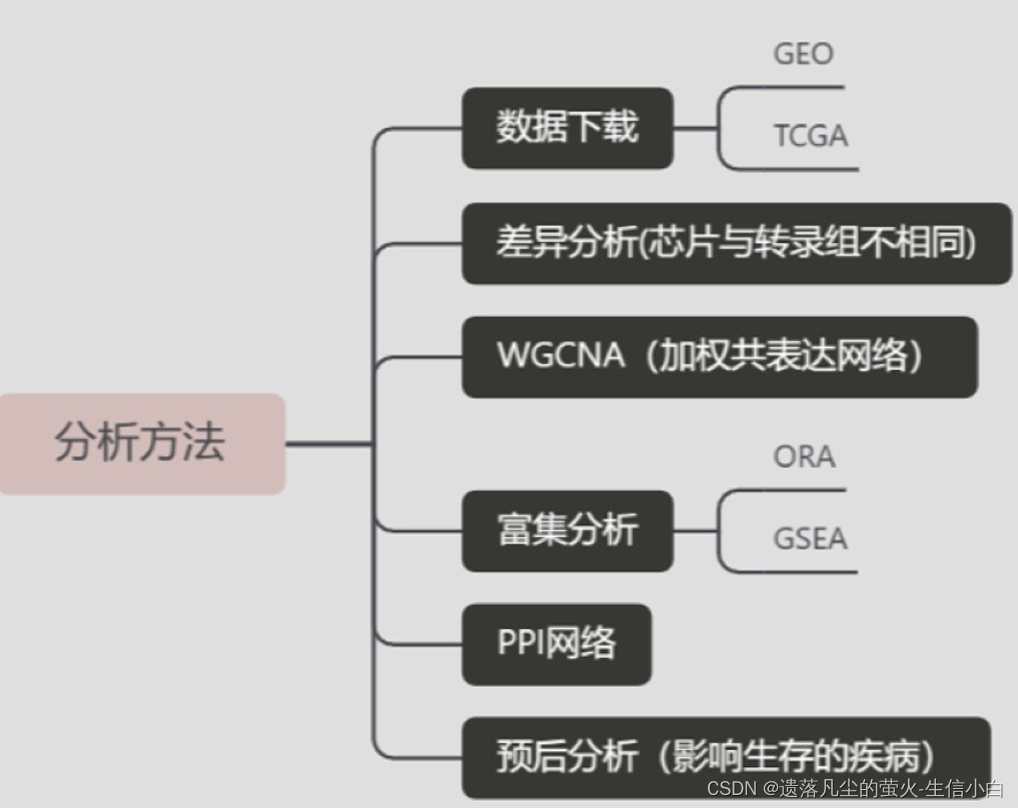
- 3.如何篩選基因(分析方法)
- 在這里插入圖片描述
- 圖表介紹
- 1.熱圖
- 2.散點圖和箱線圖
- 3.火山圖
- 4.主成分分析圖
- 如何查找數據,GEO數據庫
- 1.表達數據實驗設計
- 2.數據庫介紹
- 分析思路
- 需要整理的信息
- 芯片差異分析所需的輸入數據
- 代碼分析流程(在pipeline文件夾)
- 安裝 R 包
- 下載數據,提取表達矩陣和臨床信息
- 提取表達矩陣exp
- 檢查表達矩陣是否異常
- 提取臨床信息
- 提取芯片平臺編號,后面要根據它來找探針注釋
- Group(實驗分組)和ids(探針注釋)
- 實驗分組的設置
- 需要把Group轉換成因子,因子非常適合組織有重復值的向量,并設置參考水平,指定levels,對照組在前,處理組在后
- 探針注釋的獲取
- 運行 find_anno(gpl_number) 代碼會出現的報錯:
- 解決辦法:
- 查看ids,要求ids只能有兩列,第一列探針id,第二列symbol,并且列名都要是小寫的列名
1.圖表分析
2.GEO背景介紹及分析思路
3.代碼分析流程
4.復雜數據分析
提示:這是GEO數據挖掘的大概內容:
理論知識
廣義的基因有 6w+個,如何縮小范圍到課題相關?
1.數據從哪里來
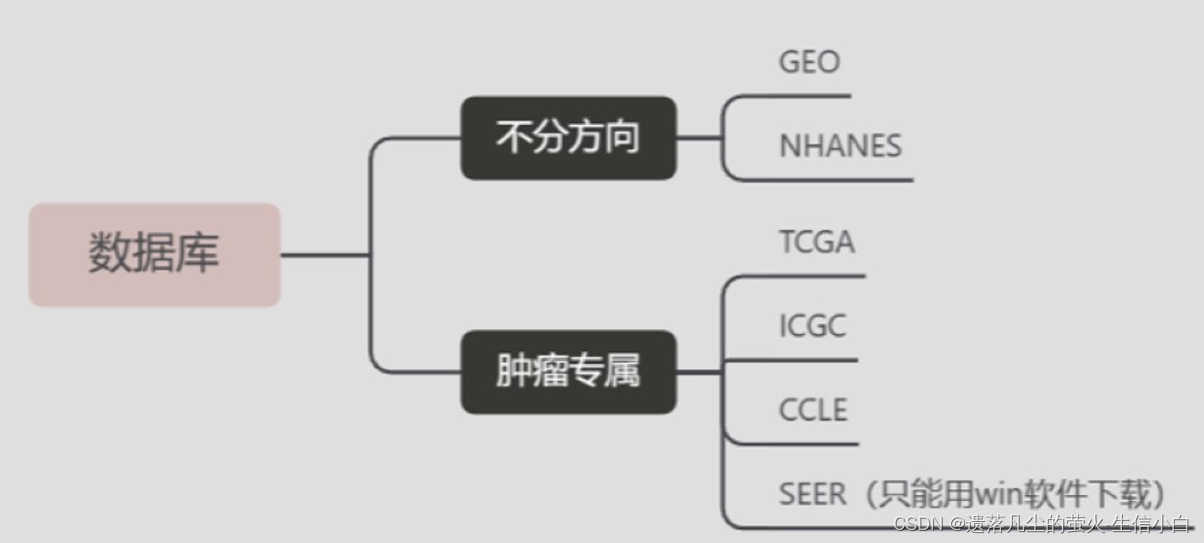
2.有什么類型的數據可挖掘
基因表達芯片
轉錄組
單細胞
突變(外顯子)、甲基化、拷貝數差異。。。
3.如何篩選基因(分析方法)
圖表介紹
1.熱圖
- 輸入數據是數值型矩陣/數據框
- 顏色的變化表示數值的大小
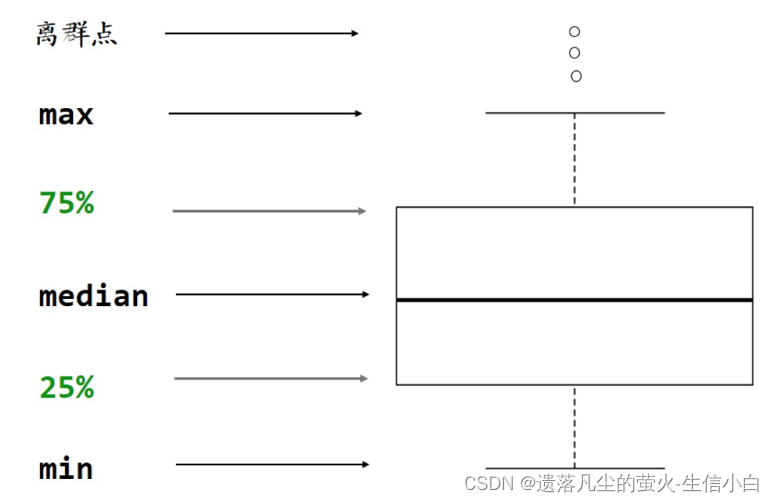
2.散點圖和箱線圖
- 橫坐標是下標/組
- 縱坐標是表達值
- 箱線圖的五條線:max、75%、median、25%、min
- 箱線圖:用于單個基因在兩組之間的表達差異
輸入數據是一個連續型向量和一個有重復值的離散型向量
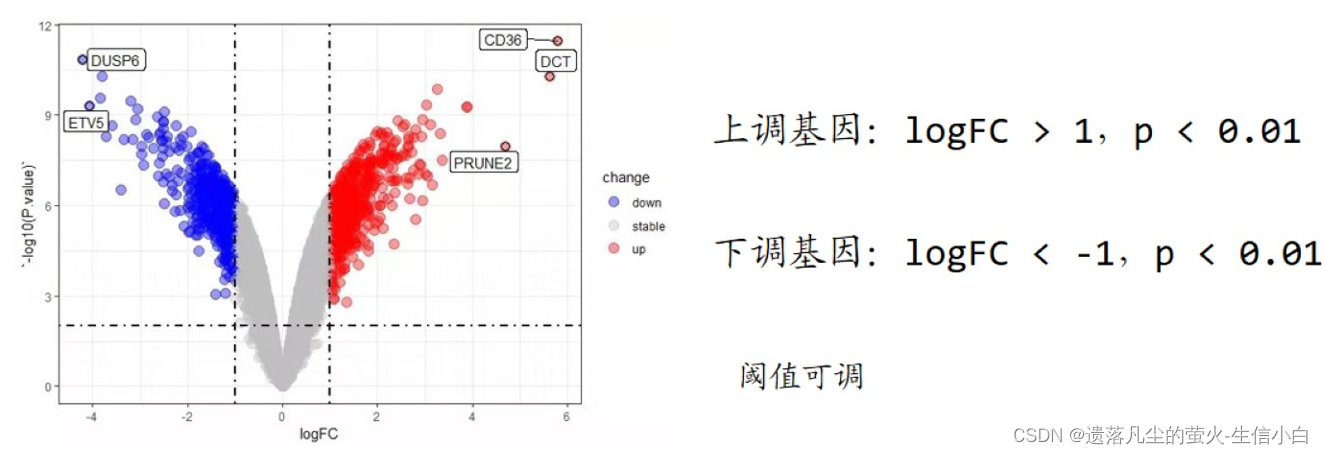
3.火山圖
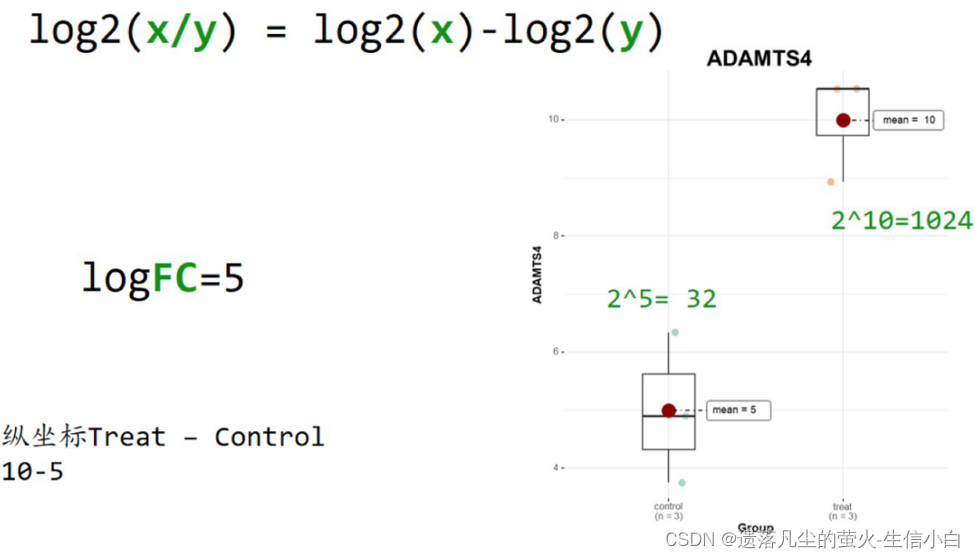
Foldchange(FC):處理組平均值/對照組平均值
log2Foldchange(logFc):Foldchange取log2
- List item
- 火山圖:差異分析,用于多個基因在兩組間的表達差異
- 橫坐標是 logFC,縱坐標是-log10(Pvalue)
- logFC: Foldchange取log2,Foldchange為處理組表達量平均值/對照組表達量平均值,實際計算時,為先取log2,然后log2(處理組)-log2(對照組),既沒有單位也沒有意義,只是一個相對值的意義
- 永遠都是處理組-對照組,而不是大的-小的
- logFC 如果太大:是不是沒取 log?頂多在 ±12 之間
- logFC > 0,treat > control,基因表達量上升
- logFC < 0,treat < control,基因表達量下降
- 通常所說的上調、下調基因是指表達量顯著上升/下降的基因(需要結合顯著性p值)
- logFC 的閾值設置是自行規定的,p 值閾值一般為 0.01,常見的 logFC 閾值有:1、2、1.2、1.5、2.2、(0.585=log2(1.5):即上升1.5倍),閾值可調。
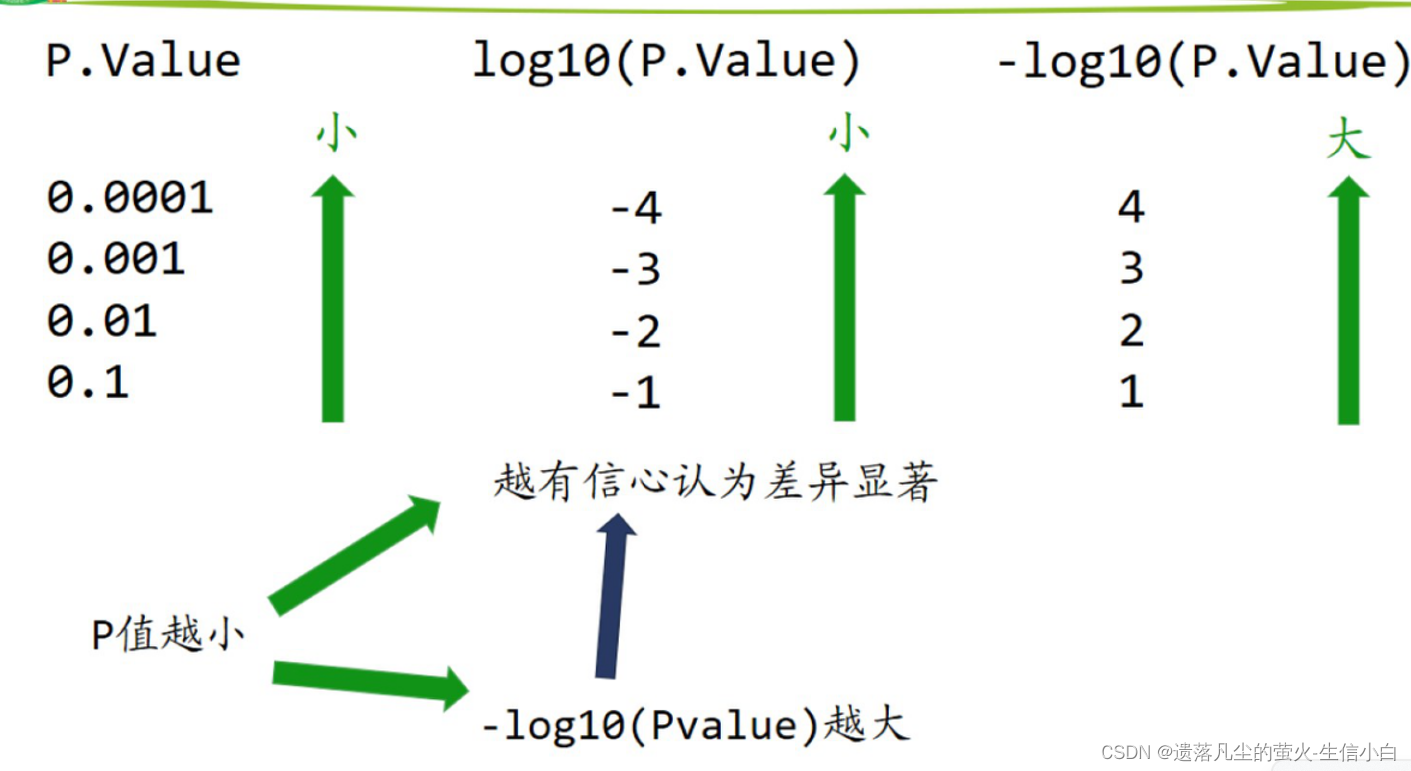
- 為什么 y 軸要取 lg?把 p 值拉到同一個數量級上,比較好畫圖
芯片差異分析的起點是一個取過log的表達矩陣(0-20),
如果拿到的是未log的矩陣(0-很大),需要自行log
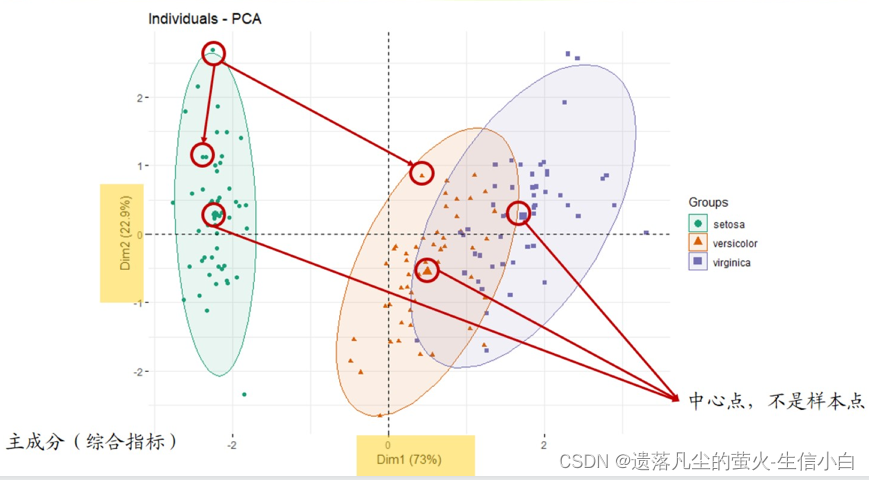
4.主成分分析圖
1.主成分分析,旨在利用降維的思想,把多指標轉化為少數幾個綜合指標(即主成分)
根據這些主成分對樣本進行聚類,代表樣本的點在坐標軸上距離越遠,說明樣本差異越大,
2. 圖上的點代表樣本(除中心點外),點與點之間的距離表示樣本與樣本的區別
3. 主成分的值沒有意義,是很多個變量降維得到的一個值,dm1和dm2是維度,dm1+dm2>90%,
4. 用處:用于“預實驗”簡單查看組間是否有差別。同一分組是否聚成一簇(組內 重復好),中心點之間是否有距離(組間 差別大)。
如何查找數據,GEO數據庫
添加鏈接描述
點進去series查找自己感興趣的數據(主要有兩種數據類型:基因表達芯片和轉錄組測序),點開樣本查看數據分布范圍,有沒有全部都在0附近,如果有大量負值,數據不正常。然后查看分組信息。

1.表達數據實驗設計
1.實驗目的:通過基因表達量數據的差異分析和富集分析來解釋生物學現象
病變組織vs健康組織
藥物處理 vs 對照組(水/DMSO)
開花前vs開花后
·動物/昆蟲不同發育期
·有色/無色果皮
高產/低產品種
有差異的材料→差異基因→找功能/找關聯→解釋差異,縮小基因范圍
2.數據庫介紹
1.GEO首頁有一個GEO2R網頁工具,它可以給相應的代碼,但是沒有判斷數據是否正常的機制,使用默認的參數,無法得到全部的默認的基因,代碼僅供參考。
2.
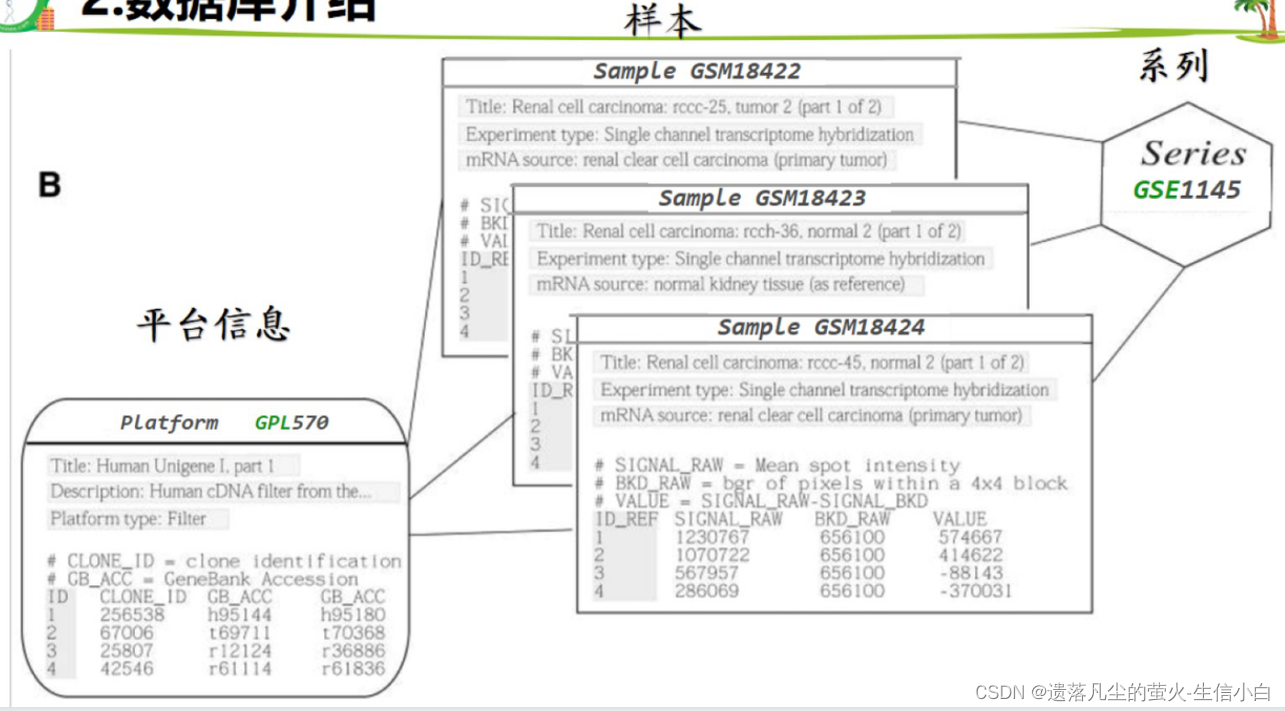
3.數據庫數據
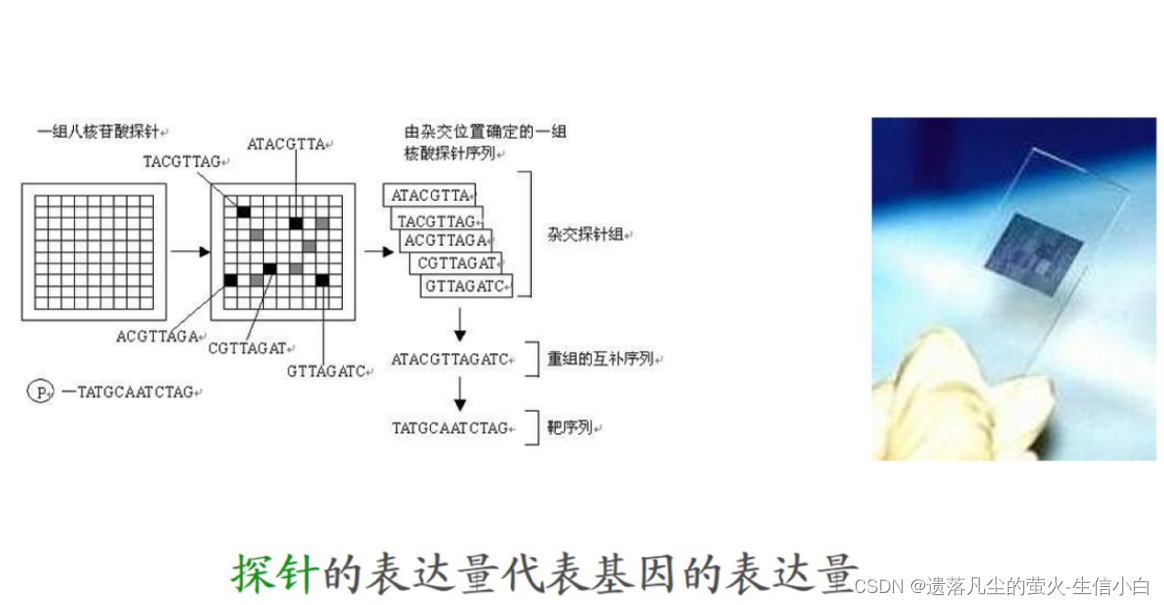
3.基因表達芯片的原理
分析思路
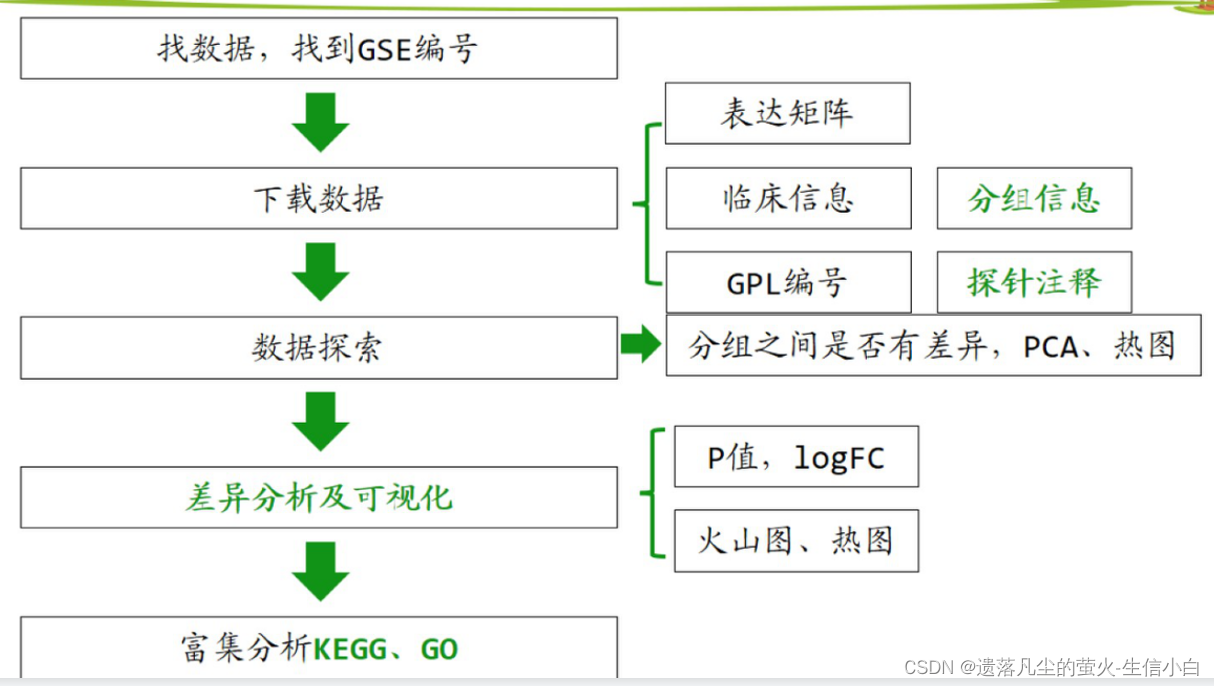
1.找數據,找到 GSE 編號
2.下載并讀取數據
3.表達矩陣
4.臨床信息 → 分組信息:需要自己整理
5.GPL 編號 → 探針注釋(每個探針屬于哪個基因,探針與基因的對應關系)
6.數據探索:分組之間是否有差異、PCA、熱圖(離散基因熱圖)
7.差異分析及可視化:limma包差異分析(P值、logFC)、畫圖(火山圖、差異基因熱圖)
8.富集分析:GO、KEGG
需要整理的信息
1.探針 id→ 基因名稱
2.GSM 樣本 ID→ 分組信息
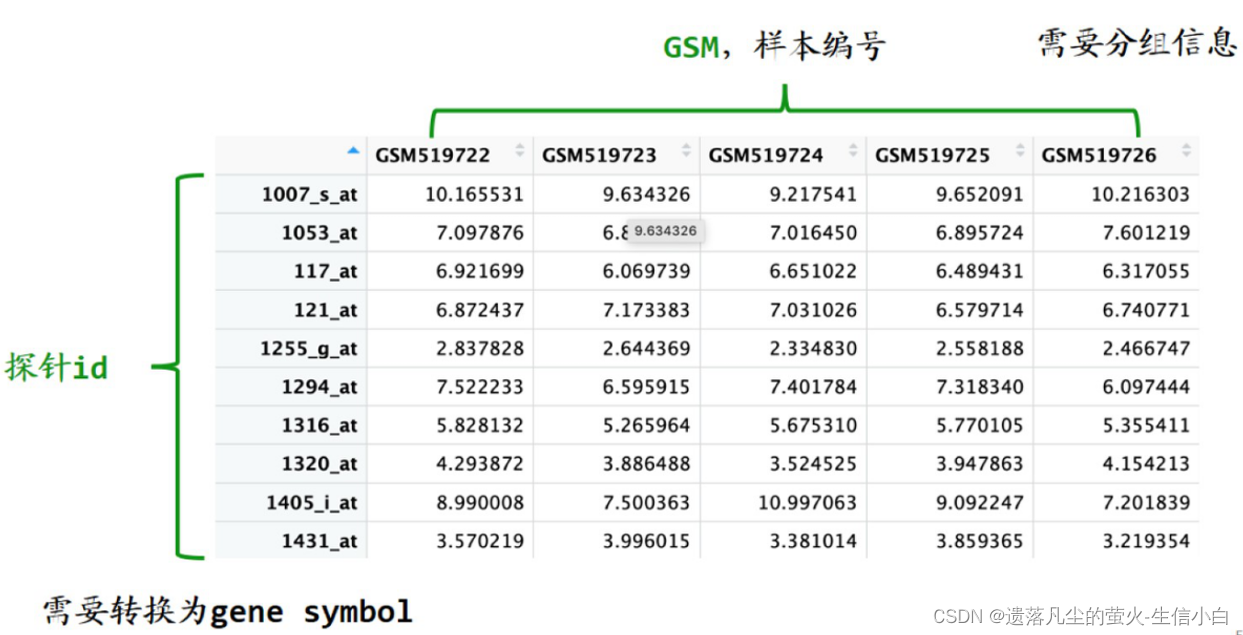
芯片差異分析所需的輸入數據

代碼分析流程(在pipeline文件夾)

安裝 R 包
00_pre_install 全選運行安裝里面的 R 包
require(pkg,character.only=T), 這個參數告訴 require() 函數,pkg 是一個字符向量。
下載數據,提取表達矩陣和臨床信息
- 下載數據,大多數情況下只要在 R 中用代碼下載即可
rm(list = ls())
#打破下載時間的限制,改前60秒,改后10w秒
options(timeout = 100000)
options(scipen = 20)#不要以科學計數法表示
#傳統下載方式
#getGEO() 函數是 GEOquery 包提供的一個工具,用于從GEO數據庫下載和導入數據集。getGEO()有則直接讀取,沒有的話就幫忙下載并讀取,在工作目錄下
library(GEOquery)
eSet = getGEO("GSE7305", destdir = '.', getGPL = F)
#destdir = '.'是指下載后放在工作目錄,getGPL是指不下載GPL文件,下載的話會變得很慢,不需要它
網速太慢,下不下來怎么辦?
1.從網頁上下載/發鏈接讓別人幫忙下,放在工作目錄里
2.試試geoChina,只能下載2019年前的表達芯片數據
library(AnnoProbe)
eSet = geoChina("GSE7305") #選擇性代替eSet = getGEO("GSE7305", destdir = '.', getGPL = F)
手動下載的方法:
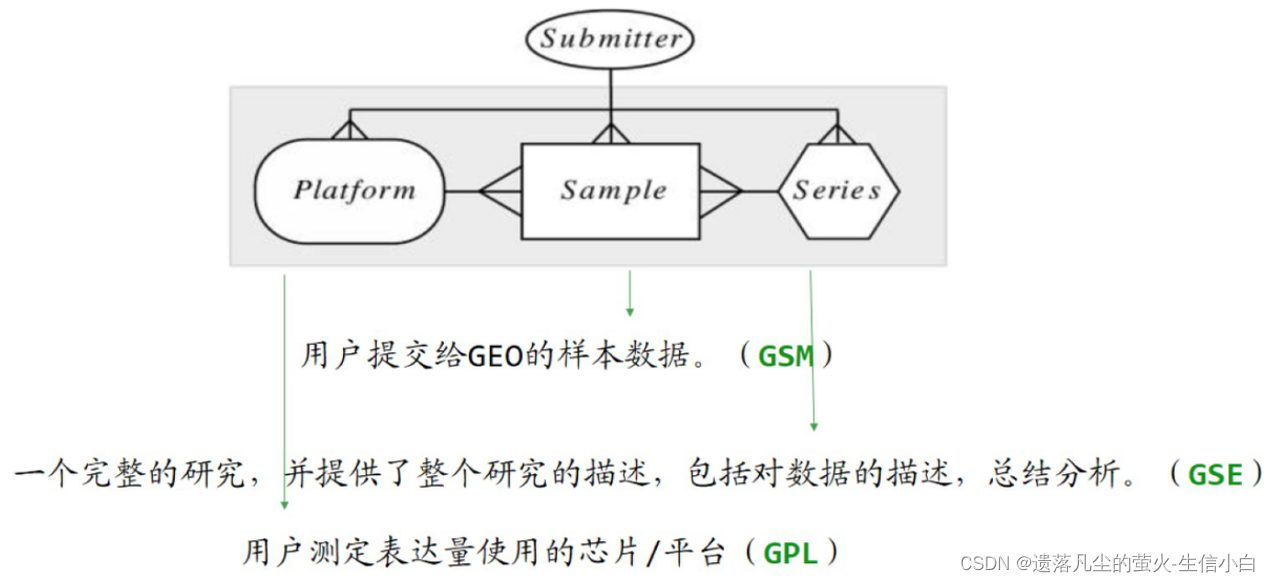
1.GEO 首頁-Series
- GSE:一個完整的研究,包括實驗設計描述、數據描述、總結分析等
- GSM:用戶提交給 GEO 的樣本數據
- GPL:指用戶用于測定表達量的芯片平臺
2.搜索需要的疾病
3.按 array 篩選檢測方法
4.點進一個 GSE
5.Series Matrix File
6.matrix.txt.gz 是表達矩陣的壓縮包(至少應該是六七百 k)
#下載下來后,研究一下這個eSet
class(eSet)
## [1] "list"
#返回列表里有幾個元素
length(eSet)
## [1] 1
#應該是1,如果有2說明是一個SuperSeries里面有兩批測序結果,如果不需要合并的話最好是分開提取,分開分析
eSet = eSet[[1]] #去掉列表數據,留下里邊數據
class(eSet)
## [1] "ExpressionSet"
## attr(,"package")
## [1] "Biobase"#ExpressionSet :數據的對象的類型
提取表達矩陣exp
exp <- exprs(eSet)
dim(exp)
#任何GEO的ExpressionSet數據都是用exprs()提取的,提取成matrix,dim() 函數用于返回一個對象的維度,這個對象可以是矩陣、數組或者數據框(data frame)。對于矩陣和數組,dim() 函數返回一個包含行數和列數的向量。
## [1] 54675 20
range(exp)#看數據范圍決定是否需要log,是否有負值,異常值
## [1] 5.020951 22011.934000
exp = log2(exp+1) #需要log才log,如果不需要取log,加上#號
#如果需要去掉一個樣本:
#exp = exp[,-3]
boxplot(exp,las = 2) # 看數據里有無異常樣本
檢查表達矩陣是否異常
正常表達矩陣
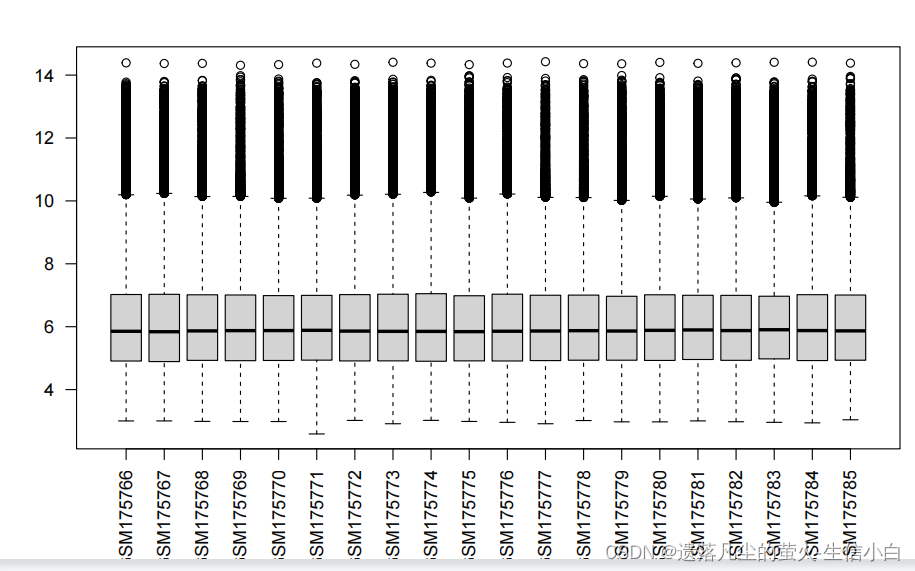
看是否有異常樣本:
數值特別大,說明沒取log
箱線圖中位數差別特別大的:
處理異常樣本:
去掉異常樣本
標準化:exp = limma::(normalizeBetweenArrays(exp))
關于表達矩陣里的負值:
1.取過log,有少量的負值:正常
2.沒取過log,有負值:不正常,錯誤數據
3.有一半負值,中位數為0:做了標準化
4.2和3情況一般棄用,非要用的話就處理原始數據
提取臨床信息
pd <- pData(eSet)
#如果需要從多個分組里取出其中兩個,多分組在這里加篩選樣本的代碼
```c
例如一個多分組:該代碼適用于所有的多分組篩選二分組
library(stringr)k1 = str_detect(pd$title,"Hep3B,control");table(k1)k2 = str_detect(pd$title,"Hep3B,control");table(k2)pd = pd[k1|k2,] #組合
#現編一個三分組
#pdKaTeX parse error: Expected 'EOF', got '#' at position 61: …es = c(6,6,8)) #?假如需要從多個分組里面取兩個分…group,“group1|group2”);table(k)
#pd = pd[k,]
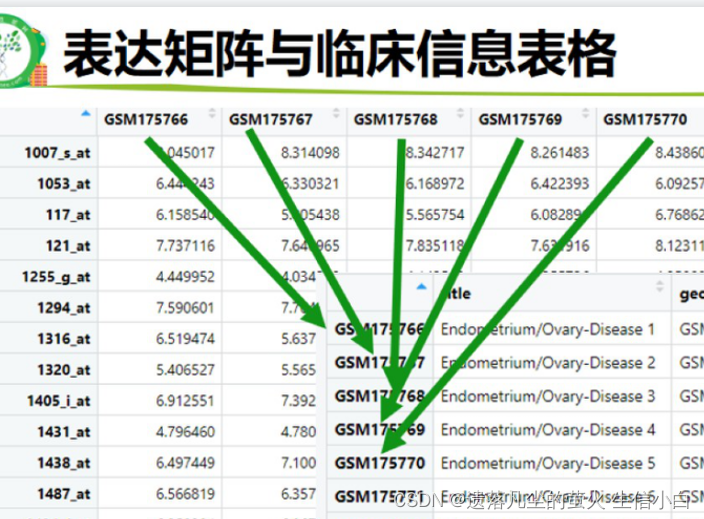
#### 讓exp列名與pd的行名順序完全一致
```c
p = identical(rownames(pd),colnames(exp));p
## [1] TRUE
#identical判斷行名和列名是否完全一致,完全相同返回T,不完全相同返回F,則運行以下代碼
if(!p) {s = intersect(rownames(pd),colnames(exp))exp = exp[,s]pd = pd[s,]
}
#s為pd里的行名和exp里的列名取交集,再使新的exp和pd變成按s順序取子集,使他們的順序完全一致
提取芯片平臺編號,后面要根據它來找探針注釋
gpl_number <- eSet@annotation;gpl_number
## [1] "GPL570"
#對象里的元素提取時用@還是$根據前面的符號,是什么就用什么
save(pd,exp,gpl_number,file = "step1output.Rdata")
#完成第一步,保存一下成Rdata
每次寫完代碼之后一定要全選運行一下,保證代碼正確且只運行了一次
附:原始數據處理的代碼,按需學習:
添加鏈接描述
Group(實驗分組)和ids(探針注釋)
# Group(實驗分組)和ids(探針注釋)
rm(list = ls())
load(file = "step1output.Rdata")
實驗分組的設置
# 1.Group----
library(stringr)
# 標準流程代碼是二分組,多分組數據的分析后面另講
# 生成Group向量的三種常規方法,三選一,選誰就把第幾個邏輯值寫成T,另外兩個為F。如果三種辦法都不適用,可以繼續往后寫else if,示例只執行第三個方法代碼
if(F){# 第一種方法,有現成的可以用來分組的列}else if(F){# 第二種方法,眼睛數,自己生成Group = rep(c("Disease","Normal"),each = 10)# rep函數的其他用法?相間、兩組的數量不同?
}else if(T){# 第三種方法,使用字符串處理的函數獲取分組k = str_detect(pd$title,"Normal");table(k)Group = ifelse(k,"Normal","Disease")
}
# **第一種方法**,有現成的可以用來分組的列
> pd$title[1] "Endometrium/Ovary-Disease 1" "Endometrium/Ovary-Disease 2" [3] "Endometrium/Ovary-Disease 3" "Endometrium/Ovary-Disease 4" [5] "Endometrium/Ovary-Disease 5" "Endometrium/Ovary-Disease 6" [7] "Endometrium/Ovary-Disease 7" "Endometrium/Ovary-Disease 8" [9] "Endometrium/Ovary-Disease 9" "Endometrium/Ovary-Disease 10"
[11] "Endometrium-Normal 1" "Endometrium-Normal 2"
[13] "Endometrium-Normal 3" "Endometrium-Normal 4"
[15] "Endometrium-Normal 5" "Endometrium-Normal 6"
[17] "Endometrium-Normal 7" "Endometrium-Normal 8"
[19] "Endometrium-Normal 9" "Endometrium-Normal 10"
#如果這一列只有"Disease","Normal"這倆單詞,不需要做任何改動,可直接運行下邊代碼,但是它除了這倆單詞還有一些額外的信息Group = pd$title
# **第二種方法**,眼睛數,自己生成
Group = rep(c("Disease","Normal"),each = 10)> rep(c("Disease","Normal"),each = 10)[1] "Disease" "Disease" "Disease" "Disease" "Disease" "Disease" "Disease" "Disease"[9] "Disease" "Disease" "Normal" "Normal" "Normal" "Normal" "Normal" "Normal"
[17] "Normal" "Normal" "Normal" "Normal"
>
#rep用法: # rep函數的其他用法?相間、兩組的數量不同?
> rep(c("Disease","Normal"),each = 10)[1] "Disease" "Disease" "Disease" "Disease" "Disease" "Disease" "Disease" "Disease"[9] "Disease" "Disease" "Normal" "Normal" "Normal" "Normal" "Normal" "Normal"
[17] "Normal" "Normal" "Normal" "Normal"
> rep(c("Disease","Normal"),times = c(8,9))[1] "Disease" "Disease" "Disease" "Disease" "Disease" "Disease" "Disease" "Disease"[9] "Normal" "Normal" "Normal" "Normal" "Normal" "Normal" "Normal" "Normal"
[17] "Normal"
> rep(c("Disease","Normal"),times = 10)[1] "Disease" "Normal" "Disease" "Normal" "Disease" "Normal" "Disease" "Normal" [9] "Disease" "Normal" "Disease" "Normal" "Disease" "Normal" "Disease" "Normal"
[17] "Disease" "Normal" "Disease" "Normal"
# **第三種方法**,匹配關鍵詞,使用字符串處理的函數獲取分組,比較萬能k = str_detect(pd$title,"Normal");table(k) #按Normal這個關鍵詞對pd$title中進行檢測,有的話就是T,沒有就是FGroup = ifelse(k,"Normal","Disease") #根據T還是F賦值group#替換成功> ifelse(k,"Normal","Disease")[1] "Disease" "Disease" "Disease" "Disease" "Disease" "Disease" "Disease" "Disease"[9] "Disease" "Disease" "Normal" "Normal" "Normal" "Normal" "Normal" "Normal"
[17] "Normal" "Normal" "Normal" "Normal" #檢查,#用pd的title列和group生成一個數據框放在一起,檢查一下分組對不對
> data.frame(pd$title,Group)pd.title Group
1 Endometrium/Ovary-Disease 1 Disease
2 Endometrium/Ovary-Disease 2 Disease
3 Endometrium/Ovary-Disease 3 Disease
4 Endometrium/Ovary-Disease 4 Disease
5 Endometrium/Ovary-Disease 5 Disease
6 Endometrium/Ovary-Disease 6 Disease
7 Endometrium/Ovary-Disease 7 Disease
8 Endometrium/Ovary-Disease 8 Disease
9 Endometrium/Ovary-Disease 9 Disease
10 Endometrium/Ovary-Disease 10 Disease
11 Endometrium-Normal 1 Normal
12 Endometrium-Normal 2 Normal
13 Endometrium-Normal 3 Normal
14 Endometrium-Normal 4 Normal
15 Endometrium-Normal 5 Normal
16 Endometrium-Normal 6 Normal
17 Endometrium-Normal 7 Normal
18 Endometrium-Normal 8 Normal
19 Endometrium-Normal 9 Normal
20 Endometrium-Normal 10 Normal
需要把Group轉換成因子,因子非常適合組織有重復值的向量,并設置參考水平,指定levels,對照組在前,處理組在后
Group = factor(Group,levels = c("Normal","Disease"))
Group##[1] Disease Disease Disease Disease Disease Disease Disease Disease Disease Disease
##[11] Normal Normal Normal Normal Normal Normal Normal Normal Normal Normal
##Levels: Normal Disease#levels(Group)
##[1] "Normal" "Disease"
##單獨提取levels,查看Group向量里都有哪些取值,這就叫因子的水平,順序對照組在前,處理組在后
探針注釋的獲取
原理:探針與基因的對應關系表格,一個數據框
需要把probe_id和基因symbol對應起來:基因組注釋
注釋來源:
1.Biocoductor的注釋包:添加鏈接描述
2.GPL頁面的表格文件解析:找探針和symbol這兩列,但不是所有文件都可以通過GPL頁面找到這兩列
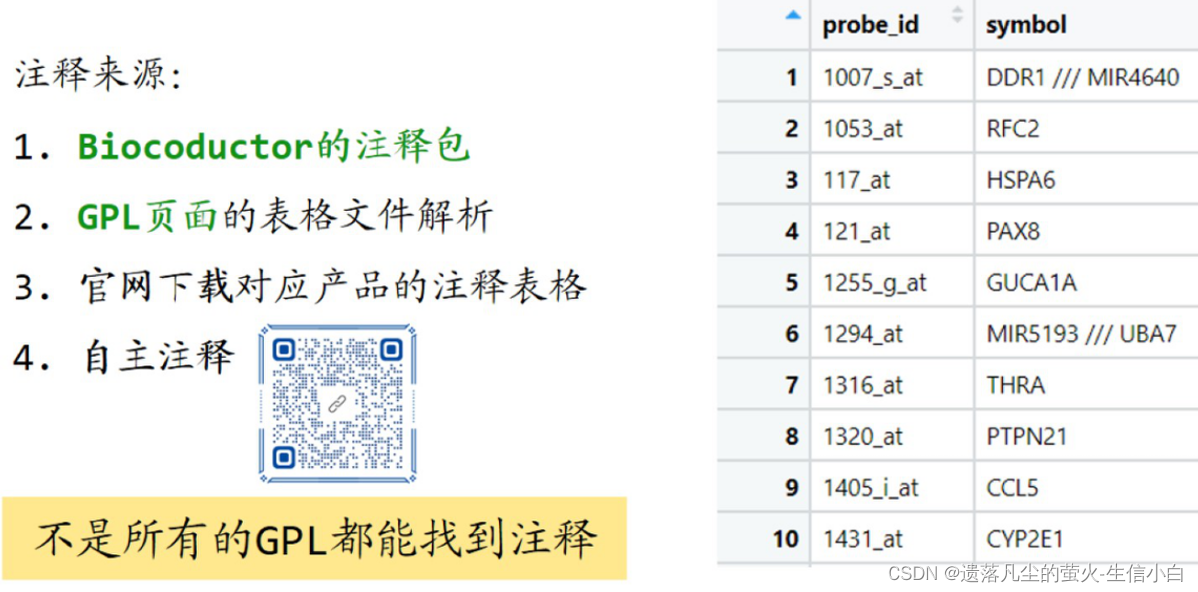
#2.探針注釋的獲取(運行的時候不同數據代碼也要改動)
#捷徑
library(tinyarray)
find_anno(gpl_number) #輔助寫出找注釋的代碼
#讀取并按列取子集的例子:GPL6887#獲取表格下載鏈接
#https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL6887&targ=self&form=text&view=data
get_gpl_txt(gpl_number)
#讀取表格
#三種方式:
a = data.table::fread("GPL6887.txt",data.table = F)
#a = rio::import("GPL6887.txt")
#a = read.delim("GPL6887.txt",check.names = F,skip = 33)
colnames(a)
ids = a[,c("ID","Symbol")]
View(ids)
#要改列名,后面的代碼適應這兩個列名
colnames(ids) = c("probe_id","symbol")
View(ids)#library(hgu133plus2.db);ids <- toTable(hgu133plus2SYMBOL)
#這是從find_anno(gpl_number)行運行結果里復制下來的代碼👆
#如果能打出代碼就不需要再管其他方法。
#如果使用復制下來的AnnoProbe::idmap('xxx')代碼發現報錯了,請注意嘗試不同的type參數
#如果顯示no annotation avliable in Bioconductor and AnnoProbe則要去GEO網頁上看GPL表格里找啦。#四種方法,方法1里找不到就從方法2找,以此類推。
#捷徑里面包含了全部的R包、一部分表格、一部分自主注釋
#方法1 BioconductorR包(最常用,已全部收入find_anno里面,不用看啦)
if(F){gpl_number #看看編號是多少#http://www.bio-info-trainee.com/1399.html #在這里搜索,找到對應的R包library(hgu133plus2.db)ls("package:hgu133plus2.db") #列出R包里都有啥ids <- toTable(hgu133plus2SYMBOL) #把R包里的注釋表格變成數據框
}
# 方法2 讀取GPL網頁的表格文件,按列取子集
##https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570
# 方法3 官網下載注釋文件并讀取
# 方法4 自主注釋,了解一下
#[添加鏈接描述](https://mp.weixin.qq.com/s/mrtjpN8yDKUdCSvSUuUwcA)
save(exp,Group,ids,file = "step2output.Rdata")運行 find_anno(gpl_number) 代碼會出現的報錯:
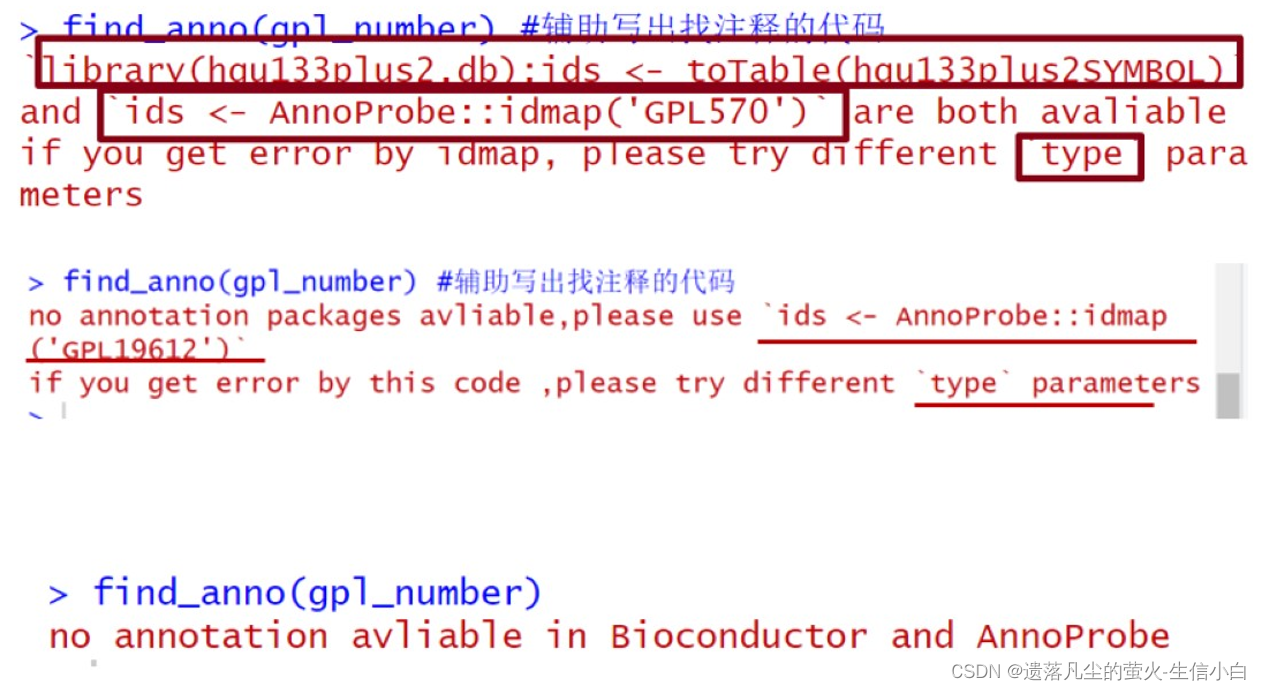
解決辦法:
方法一:
find_anno(gpl_number)
library(hgu133plus2.db);ids <- toTable(hgu133plus2SYMBOL)
方法二:
find_anno(gpl_number)
ids <- AnnoProbe::idmap('GPL570')
如果還是報錯:
?AnnoProbe::idmap #查看幫助文檔,嘗試type的三個參數,一個一個嘗試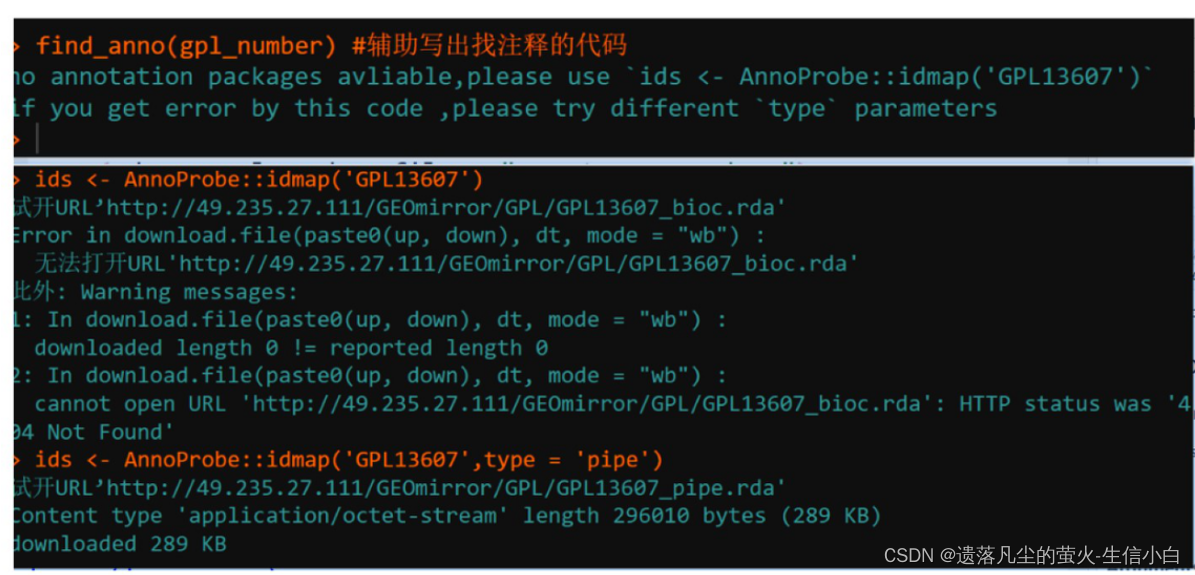
查看ids,要求ids只能有兩列,第一列探針id,第二列symbol,并且列名都要是小寫的列名
如果列名是大寫的,就更改一下
colnames(ids) = c("probe_id","symbol")








)


)
)


)



