01 背景
基因組survey以測序技術為基礎,基于小片段文庫的低深度測序,通過K-mer分析,快速獲得基因組大小、雜合度、重復序列比例等基本信息,為制定該物種的全基因組de novo測序策略提供有效依據。
jellyfish (水母) 是一個用于快速、內存高效地統計DNA中k-mer數量的工具。一個k-mer是長度為k的子字符串,統計所有這樣的子字符串的出現次數是許多DNA序列分析中的核心步驟。水母可以通過使用高效的哈希表編碼和利用“比較并交換”CPU指令來增加并行性,從而快速計數k-mer。
水母是一個命令行程序,它讀取包含DNA序列的FASTA和多FASTA文件。它以二進制格式輸出其k-mer計數,可以使用“jellyfish dump”命令轉換成人類可讀的文本格式。有關更多詳情,請參見下面的文檔。

1.1 原理
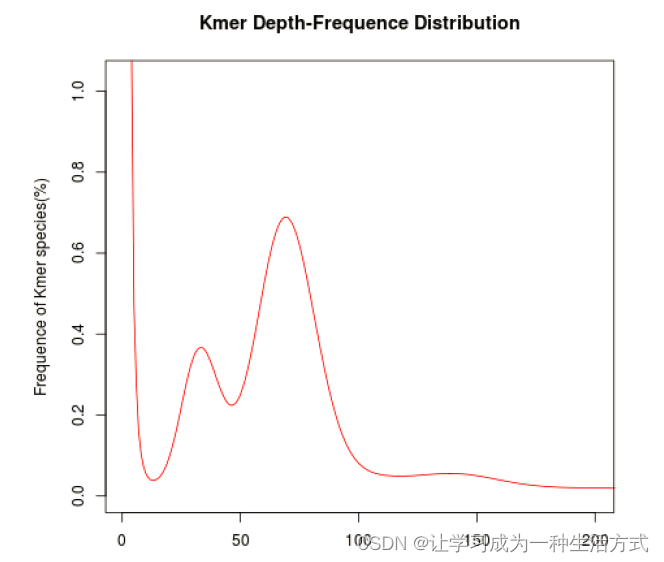
調研圖分析基于k-mer的方法,所謂k-mer是指將核酸序列以滑窗的方法分成包含k個堿基的短序列,“mer”這個單詞的來源monomeric unit,單體單元。K是常數,且一般為奇數(避免正反鏈混淆)。統計所有reads中所出現的k-mer類型及各類型k-mer的深度(或者頻率),繪制特定k-mer下不同深度k-mer片段的頻數統計圖,通常選擇K-mer分布最多的峰為主峰,從而得到基因組大小=K-mer總數/K-mer主峰深度值。
由于基因組存在雜合位點和重復序列,k-mer曲線往往不會呈現出良好的泊松分布,而是在主峰前后出現其他的峰,如果存在一定雜合度,會導致在主峰對應的橫坐標的二分之一處出現雜合峰,而一定的重復度則會在主峰對應的橫坐標的整數倍處出現重復峰。
02 參考
https://genome.umd.edu/jellyfish.html #官網
03?安裝
wget -c https://github.com/gmarcais/Jellyfish/releases/download/v2.3.1/jellyfish-2.3.1.tar.gz
tar -zxvf jellyfish-2.3.1.tar.gz
mkdir jellyfishlocation
cd jellyfish-2.3.1
./configure --prefix=/jellyfishlocation
make -j 4
make install04 使用及常用命令行
三步走,具體詳細參數一般使用缺省即可,重點關注kmer=X的值即可。
1 文件讀取,讀取多個文件
ls ~/species_name_*.clean.fq.gz | awk '{print "gzip -dc "$0 }' > generate.file
ls *.fasta.gz | xargs -n 1 echo gunzip -c > generate.file
for file in ./*.fq.gz; dogzip -dc "$file";
done > generate.file
gzip -dc 07_1.fq.gz > generate.file
gzip -dc 07_2.fq.gz >> generate.file2 k-mer的計數
#-m就是kmer的值
#計算k-mer頻率,生成sample.jf
jellyfish count -C -m 19 -s 200G -t 4 -g ./generate.file -o ./sample.jf
-m | --mer-len=<num>
使用的k-mer的長度。如果基因組大小為G,則k-mer長度選擇為: k ~= log(200G)/log(4)可選 融合二進制的輸出結果
上一步的輸出結果為二進制文件,可能輸出了多個hash文件,因此需要將這些hash文件合并成一個文件,此時用到 merge 命令。使用方法:
#jellyfish merge -o mer_counts_merged.jf hash1 hash2 ...3 對hash結果進行統計
k-mer的結果以hash的二進制文件結果給出,需要統計出k-mer總數,特異的k-mer數目,只出現過一次的kmer數,出現了最多的k-mer的數目等信息。以stats命令來運行。使用方法:
jellyfish stats hash
jellyfish stats $pre -o $pre.stat4 通過Hash結果來畫直方圖
jellyfish histo -t 10 sample.jf > sample.histo #生成k-mer頻數直方表sample.histo和k-mer直方圖
jellyfish histo -o $pre.histo $pre -t 4
?05?參考文獻
- Zimin, A. et al. The MaSuRCA genome Assembler. Bioinformatics (2013). doi:10.1093/bioinformatics/btt476
- Mar?ais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics (Oxford, England) 27, 764-70 (2011).


![贖金信[簡單]](http://pic.xiahunao.cn/贖金信[簡單])
dpdk基礎使用)





)





學習筆記)
數據操作語言)


