A near-complete assembly of an Arabidopsis thaliana?genome
擬南芥基因組的近乎完整組裝

擬南芥(Arabidopsis thaliana)基因組序列作為廣泛應用的模式物種,為植物分子生物學研究提供了巨大的推動力。在基因組序列首次發布后的20多年(Arabidopsis Genome Initiative, 2000)之后,仍然存在一些未解決的間隙區域,這些區域可能由高度重復的序列組成,如端粒、著絲粒、5S rDNA簇和含有45S rDNA的核仁組織區(NOR)。使用相對較短的測序讀取來組裝這些重復序列非常困難。對廣泛使用的TAIR10/Araport11組裝(Lamesch等,2012)進行掃描時,發現了165個間隙,涵蓋了所有五個著絲粒,并且沒有一個染色體可以從頭到尾完成。本文展示了一種高質量的組裝,包含三個無間隙的染色體和兩個只缺少NORs和NOR4末端端粒序列的染色體。通過結合長讀取Oxford Nanopore Technologies(ONT)、高保真長讀取PacBio和短讀取Illumina技術,我們獲得了一個新的133,917,231-bp的Col-0基因組組裝,命名為Col-PEK,比TAIR10/Araport11組裝大14,770,883 bp。此外,我們還填補了最近發布的兩個高質量組裝Col-CEN和Col-XJTU中大部分剩余的間隙(Naish等,2021;Wang等,2021)。在這個近乎完整的基因組組裝中,共注釋了27,629個蛋白質編碼基因,其中213個是新發現的。這些新基因中的許多位于NORs或著絲粒區域。值得注意的是,我們發現至少有145個新基因是由于之前未被識別的隱藏重復而產生的,包括串聯重復,這大大擴展了我們對近期基因重復的理解。在五個完整的著絲粒中,我們觀察到178-bp串聯衛星DNA重復序列(CEN180)的數量遠高于之前的假設。
我們整合了Nanopore ONT、PacBio HiFi和Illumina NovaSeq的讀取,用于初步組裝、修飾和去污染。隨后,我們在TAIR10的框架內將contig定位到染色體水平,然后使用HiFi contig/scaffold填補Chr4上的兩個間隙。通過填補所有間隙并定位到TAIR10,我們得到了相同的結果。最后,我們基于僅使用ONT讀取的區域修正了結構錯誤和小型拼接錯誤,并通過HiFi contig和讀取比對檢查了NORs中的潛在缺失。最終的Col-PEK組裝大小為133,917,231 bp,所有著絲粒已完成(補充方法;補充圖1)。
通過基準通用單拷貝同源基因(Supplemental Table 1)、核心真核基因映射方法評估(Supplemental Table 2)、GC-深度分析(Supplemental Figure 2)、Merqury(Rhie等,2020)和Inspector(Chen等,2021)評估(補充方法;補充表3和4)、SNP分析(Supplemental Table 5)以及使用原始Illumina過濾讀取、HiFi讀取和ONT讀取的比對(補充方法;補充圖3和4)確認了組裝質量的高水平。值得注意的是,Merqury評估表明,Col-PEK的質量明顯高于TAIR10和Col-CEN,并且與Col-XJTU的質量相當或略高(補充表3)。所有來自著絲粒區域的測序讀取都通過CEN180特異性11-mer序列得到了驗證,Merqury對五個著絲粒的評估顯示出極高的準確性,Chr2(CEN2)的錯誤率低至0(補充方法;補充表4)。我們將Col-PEK與TAIR10、Col-XJTU和Col-CEN組裝(Naish等,2021;Wang等,2021)進行了比較,發現完美的共線性(圖1A–1C;補充圖5A和6)。新的組裝添加了大約14.8 Mb的新序列,這些序列大多位于著絲粒附近或內部(圖1A;補充圖5A和7–11)。除了著絲粒之外,我們還在Chr2和Chr4的頂部臂末端分別添加了約499和約183 kb的序列(補充圖1和7–11)。序列比對表明,這些新序列包含45S rDNA亞單位(即5.8S、18S和25S rDNA)(補充表6),提示它們是NORs的一部分(Sims等,2021)。盡管長度明顯大于TAIR10(>98.55%),兩個NOR仍包含一些未完成的間隙(補充方法)。我們進一步應用覆蓋度分析來估算重復序列的拷貝數,使用Illumina讀取(Long等,2013)。估算的45S rDNA的拷貝數(>310)遠大于組裝單元的數量(約66)(補充表6),提供了NOR大小的估算值。我們還在Chr2的NOR附近鑒定了2.6 kb的端粒重復序列,而Chr4的NOR仍然缺乏端粒重復序列。總共識別出了九個端粒,大小從2.6 kb到3.6 kb不等(補充表7)。
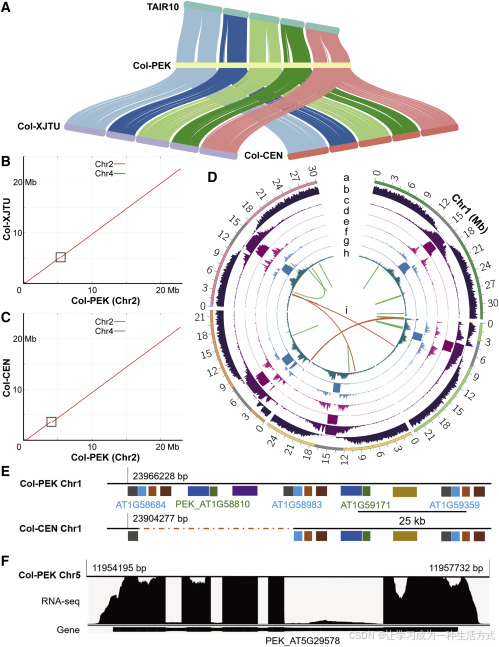
**圖1. Col-PEK組裝、注釋及應用概覽**
**(A)** 使用MUMmer比對Col-PEK、Col-XJTU、Col-CEN與TAIR10之間的序列共線性。每個共線區域表示一對一的比對關系。圖中從左到右連接的染色體分別為Chr1至Chr5。
?**(B 和 C)** Col-PEK與Col-XJTU (B) 或Col-CEN (C) 在Chr2上的共線性比較。兩個黑色框表示在Col-XJTU和Col-CEN中存在的間隙。
?**(D)** Col-PEK基因組組裝的注釋,除(h)外,其他內容均按100 kb窗口統計。
- (a) 染色體長度與著絲粒區域(灰色)以及線粒體DNA插入區域(藍色),染色體Chr1至Chr5按順時針排列;
- (b) 編碼基因數密度;
- (c) 所有重復序列密度;
- (d) CEN180單體密度;
- (e) LTR類轉座元件密度;
- (f) DNA類轉座元件密度;
- (g) 由GMATA和TRF識別的SSR與串聯重復序列的綜合密度(補充方法);
- (h) 通過Nanopolish基于ONT數據檢測的高頻CpG甲基化位點密度(以50 kb窗口);
- (i) 新識別的145個基因與其高度同源基因的共線關系。綠色線表示同一染色體上的基因,紅色線表示分布在不同染色體上的基因。
? ?**(E)** Col-PEK在Chr1上識別出Col-CEN中錯誤組裝的區域,包含一個36.0 kb的缺失序列及七個編碼基因。圖中每個矩形代表一個基因,同一面板中顏色相同表示同源基因。虛線表示Col-CEN中缺失的序列。
? ?**(F)** 一個新預測基因的示例,暫定命名為PEK_AT5G29578,位于新組裝的Chr5著絲粒中。該基因獲得RNA測序支持,最大比對深度為467×。圖底部黑色條帶表示預測的基因結構;粗條帶為預測外顯子,細條帶為預測內含子。
與最近發布的高質量組裝(Col-CEN與Col-XJTU)相比,Col-PEK組裝更完整、長度更長,并填補了多個長度超過40 kb的剩余間隙(圖1A–1C;補充圖3與6;補充表8與9;補充方法)。例如,Col-XJTU在Chr2上留下的一個108.7 kb間隙已被填補(圖1B;補充圖3A與12A)。在Col-CEN中,一個232.8 kb的不明間隙現已在Chr2的線粒體DNA插入區被識別并填補。插入后的mtDNA大小(640.5 kb)與之前熒光原位雜交估計值(618 ± 42 kb)以及Col-XJTU報告的值一致(圖1C和1D;補充圖3B、12B和13)。我們還在Col-CEN的Chr1上識別到一個36.0 kb的間隙,包含七個編碼基因(圖1E;補充圖3C、6B和12B)。這些新序列都得到了ONT和HiFi讀取的良好支持(補充圖3)。這些分析解釋了Col-PEK中Chr2、Chr4和Chr5(NOR除外)序列為何長于Col-XJTU和Col-CEN(補充表8)。另一方面,Col-PEK中的Chr1和Chr3略短于Col-XJTU,可能是由于Col-PEK中缺失了部分序列。為此,我們評估了Chr1中的一個21 kb區域(補充圖4A–4E)和Chr3中的一個11 kb區域(補充圖4F),并發現ONT通過讀取和HiFi讀取在Col-PEK斷點處有連續覆蓋,而在Col-XJTU斷點處則沒有。值得注意的是,在這些區域,Col-PEK與Col-CEN的序列完全一致(補充圖4)。
Col-PEK組裝為估算重復序列的分布提供了前所未有的機會。我們識別出26,079個簡單序列重復,總長度400,090 bp,識別出46,108個串聯重復,總長度15,470,062 bp。隨后,使用RepeatMasker(http://www.repeatmasker.org/)預測轉座元件,發現約有19,274,191 bp(占基因組的14.40%)歸屬于轉座元件。其中,LTR/Gypsy類逆轉錄轉座元件是最大的類群,占6,885,521 bp(占基因組的5.14%)。重復序列的總占比為26.58%,遠高于TAIR10的18.51%(圖1D;補充圖14;補充表8和10)。
共有27,416個蛋白質編碼基因從Araport11轉移到了Col-PEK中,總數為27,445個。剩余的基因要么位于TAIR10中錯誤組裝的區域,要么太短(3-39 bp)(補充圖15和16;補充表11;補充方法)。例如,AT3G41762在TAIR10中的26 kb錯誤組裝區域中被發現,但也有四個同源拷貝,它們被重新組裝到Col-PEK的NOR2和NOR4中(補充圖15;補充表11和12)。先前的研究也建議,TAIR10中的這個區域可能存在問題(Pucker等,2021)。值得注意的是,我們識別出145個之前未知的基因,它們與現有基因具有高度相似性(>99% DNA序列相似性)(圖1D,內圈;補充表12;補充方法)。在這些隱藏重復的基因中,70個位于兩個NORs中,47個位于前述的線粒體DNA插入區域(補充圖7–11)。根據TAIR提供的同源基因功能描述,這部分基因推測編碼線粒體呼吸途徑的蛋白質。至少56個新識別的隱藏重復基因形成了串聯重復,其中兩個或多個同源基因沿染色體相鄰排列(圖1D,內圈;補充表12)。以前也曾發現有限的隱藏基因重復現象,例如SEC10(Vuka?inovi?等,2014)(補充圖16A),而我們的發現表明這種現象更為常見(補充方法;補充表12)。不同的重復基因可能會相鄰排列。例如,Chr5中一個最近更新的區域包含兩種基因重復,一種是一個基因重復兩次,另一種是一塊三基因區域重復一次(補充表12),支持了最近的報告(Pucker等,2021)。為了進一步識別新序列中的基因,我們采用了三種獨立的方法,包括基因預測、同源搜索和參考引導的轉錄組組裝,獲得了另外68個新編碼基因,其中17個基因得到了轉錄組數據的支持(圖1F;補充圖7–11;補充表13)。這些新基因大多位于NORs和線粒體DNA插入區,一部分則分布在被著絲粒特異性組蛋白H3樣蛋白(CENH3)結合的著絲粒區域(補充圖7–11)。
由于其更高的完整性,Col-PEK在識別的非編碼RNA(ncRNA)基因數量上超過了Col-CEN和Col-XJTU。總共識別出5,959個ncRNA基因,包括3,910個編碼5S rRNA、71個編碼18S rRNA、64個編碼25S rRNA、66個編碼5.8S rRNA、648個編碼tRNA,以及1,200個編碼其他ncRNA,包括核糖開關和核糖酶(補充表6)。值得注意的是,我們的分析顯著增加了5S rRNA的數量(補充表6和8),并揭示了許多5S rRNA集中在Chr3至Chr5的著絲粒附近,并與LTR/Gypsy元素交替排列(補充圖7–11)。PacBio HiFi數據有助于填補Chr4上的間隙,并恢復易于丟失的重復序列,如5S rDNA和CEN180陣列,從而確保Col-PEK在注釋這些重復元素上的優勢(補充圖17;補充方法)。
五個完整的著絲粒為細致分析著絲粒組織提供了獨特的機會。我們共識別出66,232個著絲粒CEN180重復序列,這一數字超過了Col-CEN和Col-XJTU組裝中的數量(圖1D;補充圖7–11;補充表8)。每個著絲粒中的CEN180陣列體積從2.36 Mb到4.40 Mb不等。CENH3結合在以CEN180重復簇為中心的擴展區域,定義了功能性著絲粒。我們發現CENH3結合區域的長度大致與先前通過物理圖譜估算的著絲粒大小一致(Hosouchi等,2002;Kumekawa等,2000, 2001),并且與Col-XJTU一致,但比Col-CEN的CENH3結合區域長約1.82 Mb(補充表14)。在所有染色體中,CENH3在著絲粒核心區域富集,在LTR/Gypsy富集的區域則較少。此外,CENH3與某些CEN180子集表現出優先結合的關系。Nanopore ONT測序為檢測DNA甲基化提供了機會,這與亞硫酸鹽測序結果高度相關。我們發現NORs和5S rDNA陣列高度甲基化,而著絲粒區域的CpG甲基化水平高于染色體臂,盡管CEN180陣列相對低甲基化。此外,端粒區域則呈現低甲基化狀態(圖1D;補充圖7–11和18)。
總之,結合其他最近報告的高質量組裝(Naish等,2021;Wang等,2021),新獲得的近完整的Col-PEK組裝為擬南芥Col-0提供了一個長期期待的關鍵資源。Col-PEK的在線信息門戶,包括互動式可搜索瀏覽器以及可下載的基因組組裝和注釋文件,已上線,網址為:http://col-pek.arashare.cn/。
官網
擬南芥參考基因組_擬南芥數據庫-CSDN博客
Ensembl數據庫下載參考基因組(常見模式植物)bioinfomatics 工具37_ensembl plant數據庫-CSDN博客
http://col-pek.arashare.cn/
)
圖像與通道拼接函數-----對圖像進行幾何變換函數remap())


)
![[ linux-系統 ] 常見指令2](http://pic.xiahunao.cn/[ linux-系統 ] 常見指令2)












