WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes
WGDI:一款面向全基因組重復事件與祖先核型演化分析的易用工具集
摘要
在地球上大多數主要生物類群中,人們已檢測到全基因組復制(WGD)及其后續的核型變化。為了在基因組分析中更清晰地揭示這些復雜的多層次共線性模式,迫切需要便捷而精確的分析工具。為滿足這一需求,我們開發了WGDI(Whole-Genome Duplication Integrated analysis,全基因組復制綜合分析工具),這是一個基于Python的命令行工具,可用于全面分析多次多倍化事件及跨物種的基因組比對。
WGDI支持三種主要分析流程:多倍體推斷、基因組同源性的層級推斷以及祖先染色體核型分析。它能夠基于高質量染色體水平的基因組數據,更有效地檢測WGD事件,并深入解析與WGD相關的演化過程。值得一提的是,WGDI能提取完整的共線性區塊,并助力詳細核型進化的重建。
該工具包已在GitHub上免費開放獲取(GitHub - SunPengChuan/wgdi: WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes)。作為應用示例,WGDI成功揭示了Aquilegia coerulea(藍耬斗菜)與Vitis vinifera(葡萄)在經歷WGD之后的核型進化過程,并否定了藍耬斗菜是核心雙子葉植物異源多倍化起源的親本之一這一假說。
引言
已有確鑿證據表明,全基因組復制(WGD)或多倍化,以及伴隨而來的核型變化,在多種真核生物譜系中反復發生(Van de Peer 等,2017)。WGD 被認為是一種重要的進化過程,尤其在植物中尤為顯著(Soltis 和 Soltis,2016;Landis 等,2018)。因此,識別WGD事件、確定其發生的時間與在進化史中的位置,以及重建祖先核型,對于深入理解真核生物如何多樣化及適應不同環境至關重要(Fawcett 等,2009;Mabry 等,2020)。
目前,用于檢測WGD的主要方法分為三類:基于同義替代速率(Ks-based)、基因樹(gene tree-based)和共線性(synteny-based)的方法(Rabier 等,2014;Mabry 等,2020)。不同方法在不同數據集中的表現各有優劣(Kellis 等,2004;Hahn,2007;Vanneste 等,2013;Ruprecht 等,2017;Tiley 等,2018;Nakatani 和 McLysaght,2019;Zwaenepoel 等,2019;Zwaenepoel 和 Van de Peer,2019)。
隨著越來越多高質量染色體水平基因組的組裝發布,研究人員已開發出多種方法用于識別保留在染色體上的同源基因所形成的保守共線性區塊。早期的方法如 ADHoRe(Vandepoele 等,2002)和 DiagHunter(Cannon 等,2003),通常依賴于相鄰匹配基因對的聚類。相比之下,更新的方法使用動態規劃算法來構建成對共線基因鏈,如 ColinearScan(Wang 等,2006)、Cyntenator(R?delsperger 和 Dieterich,2010)、MCscan、MCScanX 和 JCVI(Tang 等,2017)。然而,鑒于頻繁發生的遞歸WGD以及隨后的基因組重組(通常伴隨染色體重排和大量基因丟失)使現存基因組高度復雜,這些方法在進行所有詳細后續分析方面的能力仍存在差異、不完善且耗時,具體包括生成共線基因或區塊的同源性散點圖或環圖、Ks值計算、Ks峰擬合、祖先核型演化探索以及共線基因比較等任務(Ruprecht 等,2017;Van de Peer 等,2017;Wang 等,2018)。
為促進基于高質量染色體水平基因組的WGD分析,我們開發了一個基于Python的命令行工具包——WGDI(Whole-Genome Duplication Integrated analysis,全基因組復制綜合分析)。WGDI整合了當前多數WGD相關的生物信息學分析功能,包括基因組內外的散點圖比對、共線性檢測、Ks估算與峰擬合、祖先核型演化研究及共線樹推斷。WGDI 在重建祖先核型、提取現存物種相應前體染色體方面表現出色,能夠加速WGD相關核型研究的進展。
結果
WGDI軟件包的結構
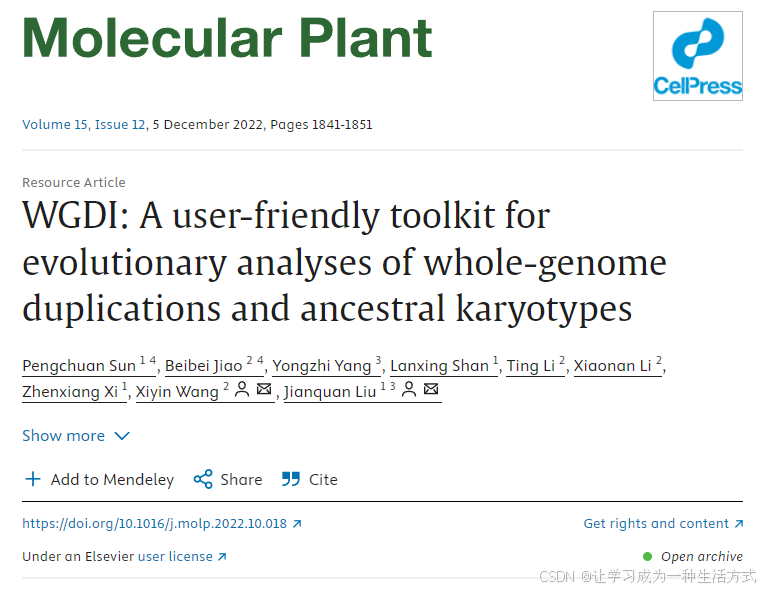
WGDI的完整源代碼可在GitHub(GitHub - SunPengChuan/wgdi: WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes)上免費獲取,并可在Windows、Linux或macOS操作系統中部署。WGDI使用Python3編寫,可通過pip或conda進行安裝。它包含多個子程序,用戶只需修改配置文件并輸入子程序名稱(例如:“wgdi -d your.conf”)即可執行相應功能。WGDI的子程序參數和功能如圖1A所示。WGDI支持三大主要工作流程(見圖1B): (1) 利用同源點圖、共線性和Ks分布以及同源基因樹進行多倍化分析與推斷; (2) 針對遞歸古多倍化事件進行基因組同源關系的分層推斷; (3) 進行亞基因組和祖先染色體核型分析及進化情景推斷。 WGDI的輸出可以包括矢量圖(如SVG格式),適合直接用于發表(見圖1C)。詳細的功能說明和參數設置可參考:Usage — WGDI 0.71 documentation。
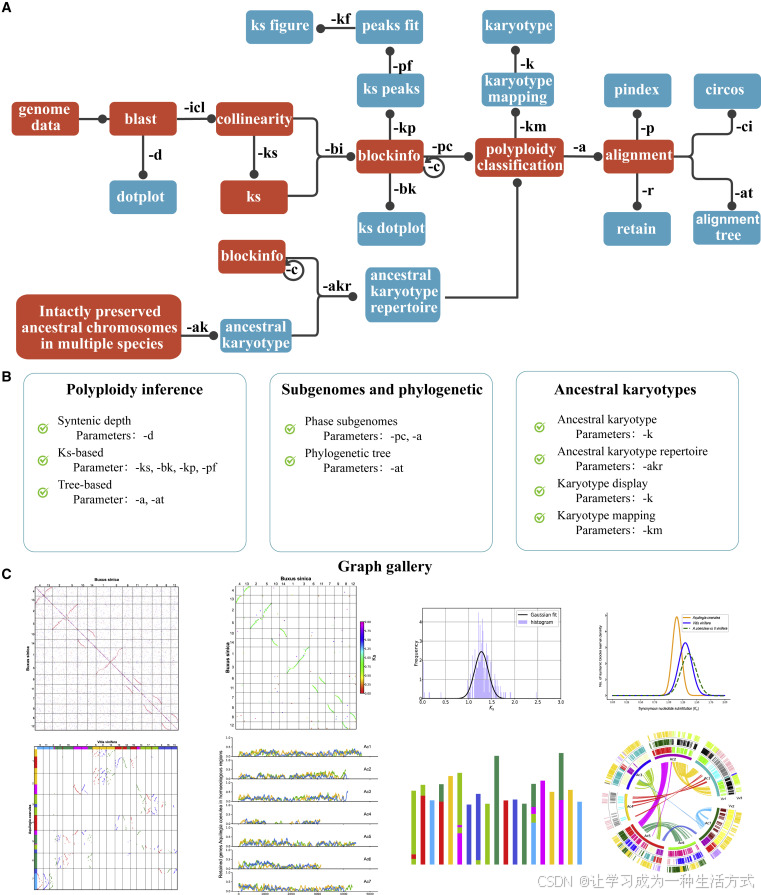
圖1. WGDI軟件包結構,展示主要組成部分及其依賴關系
(A) WGDI的多種用法以及各子程序之間的關聯關系。 (B) WGDI的三大主要工作流程及其對應的子程序。 (C) WGDI可視化輸出示例圖。更多輸出示例可直接訪問:Welcome to WGDI’s Documentation! — WGDI 0.71 documentation。
更靈敏且更準確的共線性檢測
為了評估算法性能,我們將WGDI提取的共線區與另外兩款常用工具——MCScanX和JCVI(v1.2.7)提取的結果進行了對比。三款工具使用相同的數據集進行測試,即人類(Homo sapiens)與黑猩猩(Pan troglodytes)基因組。WGDI的參數設置為:repeat_number = 10,mg = 25,25,muplite = 1,grading = 50,40,25,score = 100;MCScanX和JCVI使用默認參數。 在H. sapiens第12號染色體與P. troglodytes第13號染色體之間,WGDI、MCScanX和JCVI提取的共線區數量均為三個,但在同源塊上提取的同源基因對數量存在顯著差異(見圖2A)。WGDI提取的共線基因數量多于其他兩款工具,盡管JCVI的表現優于MCScanX。 為了進一步探究這種差異的原因,我們以一個富集了重復基因的共線區為例,展示了三款軟件的表現(見圖2B)。同源基因對在該區域內以點的形式展示,使用同源點圖的規則進行上色,共線基因對被放大并排列。WGDI、MCScanX和JCVI分別提取了49、28和40對同源基因。MCScanX忽略了這些重復基因富集區域,導致提取的同源基因對數量最少。 此外,MCScanX和JCVI提取的共線區中漏掉了一些點,甚至包括一些(用紅色標記,黑色箭頭指示)沒有重復基因的基因對。由于在共線性分析前進行了篩選,MCScanX和JCVI未能提取這些同源基因對。最終,WGDI和JCVI(默認參數)分別保留了61,117對和18,644對同源基因對。WGDI在同源矩陣中保留了更多的同源基因對,這有助于識別更長且更多的共線區。

圖2. WGDI、MCScanX與JCVI在共線性區段提取方面的性能比較
(A) 使用WGDI、MCScanX和JCVI在人類第12號染色體與黑猩猩第13號染色體之間識別局部共線性區段。三種方法提取的共線性區段中同源基因對的數量存在顯著差異。
(B) 在富含重復基因的區域提取局部共線性區段時,不同方法提取結果差異顯著。圖中以點狀表示同源基因對,放大并對齊的為共線性基因對。WGDI根據同源性將同源基因對劃分為紅色、藍色和灰色三類。圖中黑色與灰色箭頭所示位置代表被遺漏的紅點,即應被識別為共線性基因對但未被識別。
更高靈敏度的共線性檢測方法能夠保留更多同源基因對,從而提升共線性區段識別的準確性。WGDI對每個基因的同源基因對進行排序與打分,并依據顏色分類的點設定不同的搜索范圍,以此提高共線性區段的穩定性與精度(圖3A)。對于一個給定的基因,僅當其同源基因對滿足設定條件時,才被納入共線性區段。具體來說,紅、藍、灰三類點的得分依次為50、40和25,得分越高,搜索范圍越大。例如得分為50的紅點將擁有最廣的搜索范圍,而得分為25的灰點僅保留有限范圍內的基因對。對于重復基因數量超過設定參數“muplite+4”的基因,僅保留灰點類型的同源基因對。這種方式通過縮小兩端灰點的搜索范圍,降低了共線性區段邊緣識別的錯誤率,同時提升了區段長度與保留的基因對數量。
此外,由于紅點具有最高得分,共線性區段更傾向于優先保留紅點,以增強其穩定性和準確性。例如,在某一共線性區段(圖3B)中,有兩個同源點分別被標記為A和B。如果未對這些同源基因對進行排序與打分,則動態規劃算法將賦予它們相同的得分,從而無法區分其重要性。
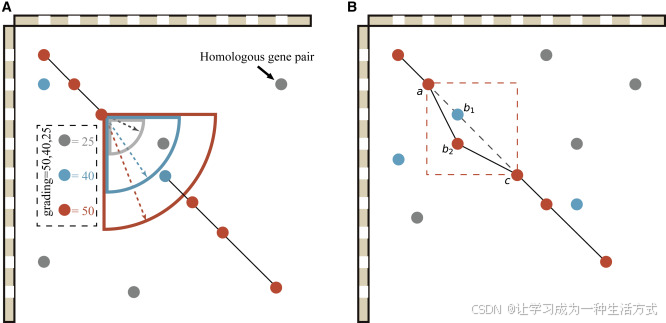
圖3. WGDI共線性檢測算法改進流程圖
(A) WGDI根據基因同源性將同源基因對劃分為紅色、藍色和灰色三類點,并賦予不同分數。不同顏色的點對應不同的搜索范圍,這種設置不僅降低了共線性區段的錯誤率,還可延長其長度。
(B) 點的不同得分也有助于增強共線性區段的穩定性。 相較之下,若未對同源基因進行排序,則無法有效識別更高同源性(如紅點)的基因對。我們使用葡萄(Vitis vinifera)、紅豆杉葉樹(Cercidiphyllum japonicum)、四合木(Tetracentron sinense)和黑胡椒(Piper nigrum)的基因組,對WGDI與其他主流軟件進行了共線性檢測性能的比較(詳見補充表1–4及補充圖1–4),結果表明WGDI在多個系統中均表現出良好的效果。
WGDI的實際應用示例
多倍化推斷分析
為展示WGDI在多倍體推斷中的應用過程,以黃瓜(Cucumis sativus)這一已知經歷多次多倍化事件的植物為例。已有研究推斷其基因組經歷過兩輪多倍化:一次為被子植物核心類群共有的三倍體化事件(WGT,或稱γ事件),另一次為較近時期的全基因組重復(WGD)(Huang 等, 2009;Wang 等, 2018)。作為參考,我們選用僅經歷過γ事件的葡萄(V. vinifera)基因組(Jaillon 等, 2007)。
多倍化分析分三步進行: 首先,算法基于共線性(同源性點圖)尋找WGD的證據。使用“-d”參數,WGDI可快速繪制V. vinifera與C. sativus之間的同源區段點圖(圖4A),以及C. sativus與V. vinifera之間的點圖(圖4B)。 縱向共線性深度為3,與所有核心雙子葉植物經歷γ事件的推斷一致;橫向共線性深度可能為2,主要由于紅色點未集中在一個共線性區段上。在主要由紅點組成的區段中,縱向共線性深度為2(圖4B)。 結合V. vinifera基因組三組同源染色體的結構,這一結果提示最大共線性深度可能為3×2=6;在紅點主導的區段中,其橫向共線性深度為1,對應最大深度為3×1=3。 綜合已有的多倍體事件推斷和共線性深度信息,可以構建一幅系統進化樹(圖4F)。由此可見,WGDI生成的同源點圖可適應大多數關于WGD事件的推斷模型。
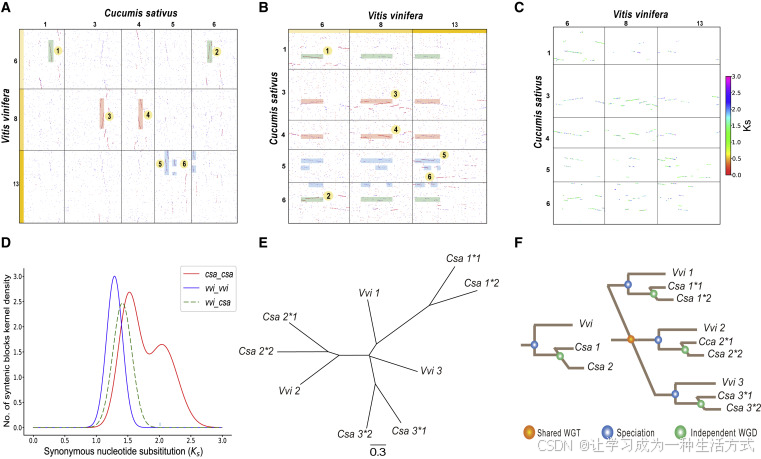
圖 4. 以黃瓜(*Cucumis sativus*)和葡萄(*Vitis vinifera*)為例的多倍體推斷
(A) 所選葡萄與黃瓜染色體之間同源關系的點陣圖。葡萄的6、8和13號染色體是由核心真雙子葉植物全基因組三倍化產生的同源三聯體。 (B) 所選黃瓜與葡萄染色體之間的同源關系點陣圖。圖中用矩形標出了外旁系區域或次級匹配區域。 (C) Ks 點陣圖,其中基于 Ks 值對共線性區塊進行識別并著色。 (D) Ks 分布圖。 (E) 兩個物種的基因系統發育樹。 (F) 使用 ASTRAL-III 構建的比對樹推斷系統發育關系。
第二步使用 Ks 分布來驗證全基因組加倍(WGD)的發生及其時間。使用 “-bk” 程序,可以直觀地可視化共線性基因對的 Ks 值(圖 4C)。同一共線區塊中的基因對 Ks 值波動較小,可以使用其中位數代表該區塊的 Ks 分布。不同區塊的 Ks 值差異很大,尤其是在區塊來源于不同多倍化事件的情況下。通過結合參數和 Ks 分布,可以輕松區分這些區塊,并使用 “-pf” 程序輸出擬合函數及其最優擬合。目前,許多研究者仍使用高斯混合模型或類似方法擬合多個多倍化事件。但由于高斯混合模型容易過擬合,得到的 Ks 峰可能波動大、不準確(Zwaenepoel 和 Van de Peer,2019)。可使用 “-kf” 程序顯示單一或多個物種間的 Ks 分布。可以看出,葡萄與黃瓜共享的三倍化事件的 Ks 峰差異很大,甚至葡萄和黃瓜的分化事件的 Ks 峰比黃瓜最近一次 WGD 的 Ks 峰還要小(圖 4D)。由于物種的進化速率差異較大,即使是同一次 WGD,其 Ks 峰也可能有很大不同。因此,必要時需對 Ks 峰進行校正,然后推斷多倍化時間(Wang 等,2017,2018;Yang 等,2020)。與基于聚類得到的旁系基因對(如 OrthoMCL 使用的方法)相比,WGDI 推斷的 Ks 分布可大大減少串聯重復或其他非共線性旁系基因帶來的影響。
最后,使用基于基因樹的方法驗證多倍化推斷。在本研究中,使用 “-a” 和 “-at” 程序,獲得了葡萄和黃瓜的同源基因列表,并通過 ASTRAL-III v5.7.7 構建的 1138 個基因樹推斷其系統發育關系(Zhang 等,2018)(圖 4E)。其中有 92 個基因樹至少包含三個相同的黃瓜基因,占所有基因樹的 8.08%。在一些缺失多個同源基因的基因樹中,其結構仍支持黃瓜經歷了一次較新的 WGD 的假設。總之,WGDI 將三種主要的 WGD 檢測方法結合在一起,能夠相互驗證,幫助研究人員準確、高效地推斷多倍化事件。
WGDI 在多層次基因組同源推理中的應用
花生(Arachis hypogaea)是一種異源多倍體,已被證明來源于兩個祖先物種——Arachis duranensis 和 Arachis ipaensis 的雜交(Bertioli 等,2016, 2019;Zhuang 等,2019)。在此,我們以花生為例,說明如何通過以下步驟在異源多倍體中推斷基因組同源關系:
第一步,使用同源點陣圖和 Ks 點陣圖對基因組同源性進行分層推理。通過 “-d” 和 “-bk” 程序輸出結果可以看出,花生染色體1至10來自 A. duranensis,染色體11至20來自 A. ipaensis。然而,染色體3和13之間顯然發生了易位(圖 5A)。通過評估不同多倍化事件保留的共線區塊的 Ks 值、區塊標識等差異,可以使用 “-a” 程序輕松獲得三種基因組間的同源基因列表(圖 5B)。
第二步,使用比對樹進行進一步的分層推理。通過 “-at” 程序,使用來自經歷了染色體易位的花生3號染色體、A. duranensis 和 A. ipaensis 染色體的同源基因列表構建了2376棵基因樹,并以滑動窗口形式顯示所有可能的樹結構得分(圖 5C)。其中1597棵支持花生起源于 A. duranensis,624棵支持起源于 A. ipaensis,與所推斷的染色體易位一致。
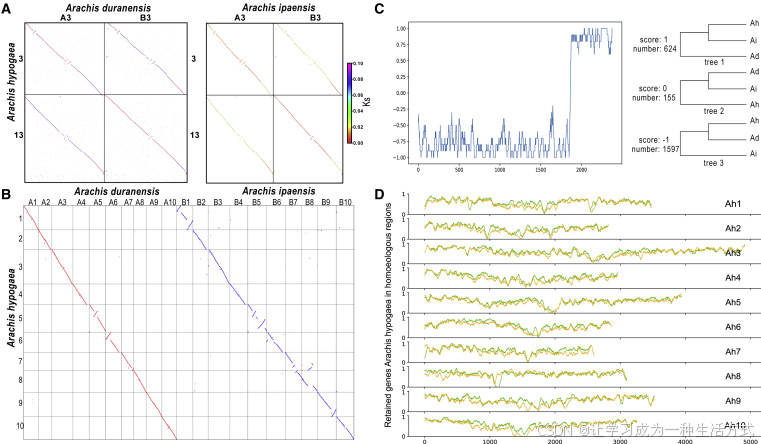
圖5. 以落花生(*Arachis hypogaea*)為例進行基因組同源性的分層推斷
(A) 同源性點圖和Ks值點圖,比較選定的落花生(A. hypogaea)、杜蘭花生(A. duranensis)和伊帕花生(A. ipaensis)的染色體。圖中展示了落花生的染色體3和13,它們是由共有的雙子葉植物六倍化事件產生的同源三聯體,其匹配的染色體為杜蘭花生的染色體3和伊帕花生的染色體4。
(B) 以杜蘭花生為參考基因組,對落花生進行的比對結果。
(C) 在落花生染色體3的同源性列表中,所有可能的系統發育樹結構的分布。
(D) 滑動窗口中落花生同源區域1(橙色)和同源區域組2(綠色)的基因保留率。
最后,通過“–r”和“–p”程序,分別展示了該異源四倍體中的基因保留情況和亞基因組分化情況。在異源四倍體落花生中,來自杜蘭花生亞基因組的基因保留數量多于來自伊帕花生亞基因組的(見圖5D)。此外,兩套亞基因組或祖先二倍體基因組之間的分化(P指數)估計為0.86,說明它們存在顯著差異,符合典型異源四倍體的特征。因此,WGDI使用多種方法,基于基因組同源性進行異源多倍化的分層推斷,并生成由基因共線性支持的、與異源多倍體物種形成相關的同源基因的分層列表。
WGDI用于追蹤祖先染色體核型的應用
葡萄(Vitis vinifera)的染色體核型廣泛用作核心雙子葉植物核型分析的參考。此前的核型分析表明,葡萄的祖先染色體發生過兩次融合和四次斷裂(Murat等,2015)。根據核型變化,有人推測耬斗菜(Aquilegia coerulea,毛茛科)是所有核心雙子葉植物的親本祖先之一,可能是一個古老的異源六倍體(Akoz和Nordborg,2019)。然而,我們使用“–km”程序,基于祖先雙子葉植物核型(AEK)獲得了葡萄和耬斗菜的核型。與之前的核型結果相比,我們在之前的空白區域恢復出了明顯的共線性區塊(見圖6A)。例如,耬斗菜染色體2和染色體3的末端以及染色體5的起始部分對應于葡萄染色體4的中間區域(圖6A),該區域是未注釋區域(空白區域,參見Akoz和Nordborg,2020年的圖6)。如果核型演化基于這些結果,可能會造成誤導。
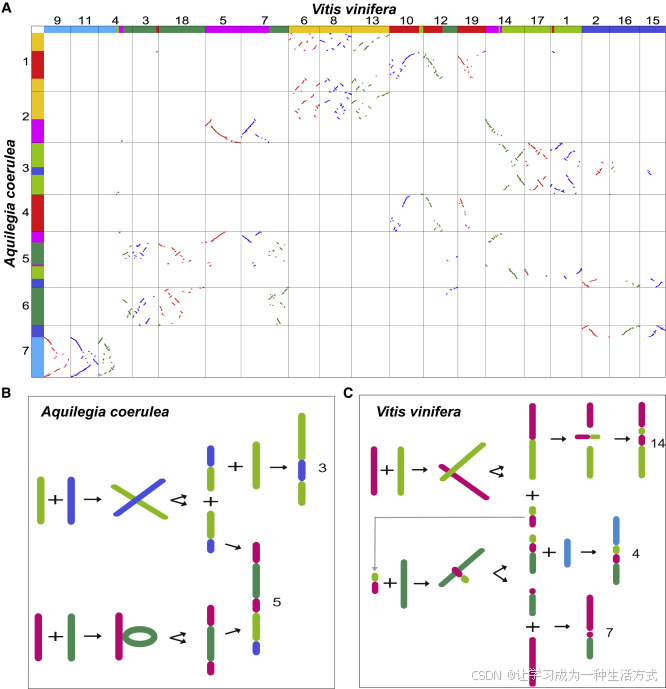
圖6. 葡萄(*Vitis vinifera*)染色體4、7和14與耬斗菜(*Aquilegia coerulea*)染色體3和5的核型演化
(A) 選定的耬斗菜和葡萄染色體之間的同源性點圖。圖中七種不同顏色代表七條祖先單倍體染色體。
(B) 耬斗菜染色體3和5的構建過程。
(C) 葡萄染色體4、7和14的構建過程。
染色體數目減少與B染色體模型(Wang等,2015;Wang和Wang,2020)為理解真核生物染色體數目及其重排的演化提供了新方法。通過該模型,發現葡萄的祖先核型比以前推測的更加復雜但也更清晰。例如,粉紅色和草綠色染色體在靠近各自一端的地方發生了交叉,形成了兩個新染色體(圖6C)。兩段較長的片段融合成葡萄染色體14,之后在該染色體內部發生了倒位。兩段較短的片段并未消失,而是與其他染色體融合,最終形成葡萄染色體4。粉紅色和綠色染色體也發生了類似的融合,最終形成了現在的染色體7和4。
然而,耬斗菜的核型與葡萄完全不同。綠色染色體插入粉紅色染色體,草綠色染色體與藍色染色體發生交叉,隨后分別演化為現在的染色體5和3(圖6B)。因此,耬斗菜和葡萄的核型(參見Akoz和Nordborg,2020)并不能作為支持“耬斗菜基因組揭示核心雙子葉植物雜交起源”的證據。與之前的研究結果相比,WGDI揭示的新結論可能更準確地反映了祖先染色體重排的動態過程。
討論
基因共線性的分析可為多倍化研究提供有力的見解,多倍化在許多物種和系統的形成與演化中發揮了重要作用。WGDI尤其適用于識別頻繁發生的多倍化事件,尤其是在高質量的染色體級別基因組基礎上。該工具提供的分層推斷和與WGD事件相關的基因共線性推斷,有助于厘清因重復多倍化而復雜化的植物系統發育關系和核型演化。
WGDI在共線性檢測方面的表現(見補充表1–4和補充圖1–4)與以往工具(Wang等,2006, 2012;Tang等,2017)相當。
此外,WGDI在重建現存物種的祖先染色體核型方面也非常有效和實用。WGD會導致快速的基因組重組和結構變異,從而在新物種中產生新的染色體核型。理解這種核型演化對于評估某些系統位置尚存爭議的譜系以及推測滅絕物種的基因組結構具有重要意義(Akoz和Nordborg,2020)。然而,分裂與融合模型(Salse等,2008;Murat等,2010)無法解釋祖先基因組的染色體是如何演化為現今核型的(Wang等,2015)。例如,被推測為所有被子植物、單子葉植物和核心雙子葉植物祖先核型(Murat等,2017)中,存在大量未注釋(空白)區域。在核型演化過程中,共線性或共線性斷裂是推斷祖先核型的重要特征。相比簡單的分裂–融合假說,植物與動物都需要更詳細的染色體演化圖譜(Akoz和Nordborg,2020)。WGDI能夠提取更完整的共線性區塊,有助于從祖先染色體中重建詳細的核型演化過程。
WGDI采用了基于動態規劃的共線性提取算法,并集成了多種用于可視化和分析的計算程序。對分層和事件相關共線性的推斷,有助于厘清被反復多倍化干擾的真實系統發育關系。WGDI可通過GitHub(GitHub - SunPengChuan/wgdi: WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes)免費獲取,并支持conda環境和bioconda平臺(Grüning等,2018),實現兼容、易于安裝和升級的工具使用體驗。
)





的失配誤差校正處理(以4片1GSPS采樣率的12bitADC交織為例講解))
之管道1【匿名管道】)




VTK C++開發示例 ---圖片轉3D)





:分析器)
)