最近,用Python腳本提取,在基因號已知,位置已知條件下,相對應位置的基因序列時發現,這樣很簡單但是很實用的腳本,在網上卻比較難找。而且,能被找到的腳本,相對于具有初級編程能力的人而言,有點難。本人寫了相對于初學者同樣很簡單腳本分享給大家。
首先,我將fa文件處理為單行(嫌麻煩,沒有寫成scaffold_x一行,序列一行的樣子,如圖三),將下面的序列處理(圖一):

(補充)經過:
import re
fr=open(r'F:desktopcorrelxxx.fa','r')
fw=open(r'F:desktopcorrelxxx_use.fa','w')
line=fr.read()
r=line.replace('n','')
s=re.sub('>','n>',r)
fw.write(s)
fr.close()
fw.close()
得到(圖二):

當然你如果不嫌麻煩也可以處理成(圖三):

假設我含有位置信息源文件(圖四):

第一列為基因號,最后一列為基因在fa文件中的位置信息;
本人采用圖二的形式,具體腳本(腳本一);
#author:Wang Binzhong
# -*- coding:utf-8 -*-
fr=open(r'F:desktopCX.txt','r')#讀取含有位置信息的文件
fa=open(r'F:desktopxxxx.fa','r')#讀取處理好的基因序列文件
fw_1=open(r'F:desktopfa_3.txt','w')#寫入
line_cr=fr.readlines()
line_fa=fa.readlines()
for eachline in line_cr:
sp=eachline.strip().split('t')
title_1=eachline.find('scaffold')
start_1=eachline.find(':',title_1)+1
end_1=eachline.find('-',start_1)
d_1=eachline[title_1:start_1-1].strip()#scaffold名稱
d_2=eachline[start_1:end_1].strip()#首位的位置
d_3=eachline[end_1+1:].strip()#末尾的位置
for each_seq in line_fa:
if d_1 == each_seq[:int(len(d_1))+5].strip('ATGC'):#如果對應的名稱在行中,就可以用以下的規則寫入文本
fw_1.write(sp[0]+'t'+each_seq[len(d_1)+int(d_2):len(d_1)+int(d_3)].strip()+'n')#改為:fw_1.write('>'+sp[0]+'n'+each_seq[len(d_1)+int(d_2):len(d_1)+int(d_3)].strip()+'n')可以省略第二步(腳本二),一步完成
break
fr.close()
fa.close()
fw_1.close()
表頭沒有'>',同時也沒有換行處理,所以需要繼續處理(圖五):
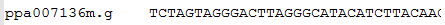
沒有寫連續的腳本,重新寫了一個(腳本二):
import re
fr=open(r'F:desktopfa_3.txt','r')
fw=open(r'F:desktopfa_4.fa','w')
line_fr=fr.readlines()
s_1=''
for eachline in line_fr:
s_1=re.sub('t','n',eachline)
fw.write(re.sub('pp','>pp',s_1))
fr.close()
fw.close()
最終得到:

程序比較簡單,Python初學者都可以懂。當然,如果有錯誤的地方可以留言指出,
 希望能為需要的同學提供幫助。這個程序只是針對于正鏈的
希望能為需要的同學提供幫助。這個程序只是針對于正鏈的
注:之前寫的出現了一個bug,經過修改后發布成功提取序列,希望對各位有幫助,有用的話可以引用。
鑒于某些人盜版,轉載請注明網址。
轉載本文請聯系原作者獲取授權,同時請注明本文來自王彬忠科學網博客。
鏈接地址:http://blog.sciencenet.cn/blog-783116-801490.html



深入理解BIO、NIO、AIO)





)
運算符)

.doc)
方法實例)





用法及代碼示例)