吳恩達神經網絡1-2-2
預測溶解度 (Predicting Solubility)
相關資料 (Related Material)
Jupyter Notebook for the article
Jupyter Notebook的文章
Drug Discovery with Graph Neural Networks — part 2
圖神經網絡進行藥物發現-第2部分
Introduction to Cheminformatics
化學信息學導論
Deep learning on graphs: successes, challenges, and next steps (article by prof Michael Bronstein)
圖上的深度學習:成功,挑戰和下一步 (邁克爾·布朗斯坦教授的文章)
Towards Explainable Graph Neural Networks
走向可解釋的圖形神經網絡
目錄 (Table of Contents)
- Introduction 介紹
- A Special Chemistry Between Drug Development and Machine Learning 藥物開發與機器學習之間的特殊化學
- Why Molecular Solubility is Important 為什么分子溶解度很重要
- Approaching the Problem with Graph Neural Networks 圖神經網絡解決問題
- Hands-on Part with Deepchem Deepchem的動手部分
- About Me 關于我
介紹 (Introduction)
This article is a mix of theory behind drug discovery, graph neural networks and a practical part of Deepchem library. The first part will discuss potential applications of machine learning in drug development and then explain what molecular features might prove useful for the graph neural network model. We then dive into coding part and create a GNN model that can predict the solubility of a molecule. Let’s get started!
本文綜合了藥物發現,圖形神經網絡和Deepchem庫的實際部分的理論知識。 第一部分將討論機器學習在藥物開發中的潛在應用,然后解釋什么分子特征可能對圖神經網絡模型有用。 然后,我們深入編碼部分,并創建可以預測分子溶解度的GNN模型。 讓我們開始吧!
藥物開發與機器學習之間的特殊化學 (A Special Chemistry between Drug Development and Machine Learning)
Drug development is a time-consuming process which might take decades to approve the final version of the drug [1]. It starts from the initial stage of drug discovery where it identifies certain groups of molecules that are likely to become a drug. Then, it goes through several steps to eliminate unsuitable molecules and finally tests them in real life. Important features that we look at during the drug discovery stage are ADME (Absorption, Distribution, Metabolism, and Excretion) properties. We can say that drug discovery is an optimization problem where we predict the ADME properties and choose those molecules that might increase the likelihood of developing a safe drug [2]. Highly efficient computational methods that find molecules with desirable properties speed up the drug development process and give a competitive advantage over other R&D companies.
藥物開發是一個耗時的過程,可能需要數十年才能批準該藥物的最終版本[1]。 它從藥物發現的初始階段開始,在此階段它可以識別可能成為藥物的某些分子組。 然后,它通過幾個步驟來消除不合適的分子,并最終在現實生活中對其進行測試。 重要的特征,我們在藥物開發階段看是ADME(A bsorption,d istribution, 男 etabolism和E xcretion)性能。 可以說,藥物發現是一個優化問題,我們可以預測ADME特性并選擇可能增加開發安全藥物可能性的分子[2]。 查找具有所需特性的分子的高效計算方法可加快藥物開發過程,并提供優于其他研發公司的競爭優勢。
It was only a matter of time before machine learning was applied to the drug discovery. This allowed to process molecular datasets with a speed and precision that had not been seen before [3]. However, to make the molecular structures applicable to machine learning, many complicated preprocessing steps have to be performed such as converting 3D molecular structures to 1D fingerprint vectors, or extracting numerical features from specific atoms in a molecule.
將機器學習應用于藥物發現只是時間問題。 這樣可以以前所未有的速度和精度處理分子數據集[3]。 但是,要使分子結構適用于機器學習,必須執行許多復雜的預處理步驟,例如將3D分子結構轉換為1D指紋矢量 ,或從分子中的特定原子提取數值特征。
為什么分子溶解度很重要 (Why Molecular Solubility is Important)
One of the ADME properties, absorption, determines whether the drug can reach efficiently the patient’s bloodstream. One of the factors behind the absorption is aqueous solubility, i.e. whether a certain substance is soluble in water. If we are able to predict the solubility, we can also get a good indication of the absorption property of the drug.
ADME的特性之一就是吸收,它決定藥物是否可以有效地到達患者的血液中。 吸收背后的因素之一是水溶性,即某種物質是否可溶于水。 如果我們能夠預測溶解度,我們也可以很好地表明藥物的吸收特性。
圖神經網絡解決問題 (Approaching the Problem with Graph Neural Networks)
To apply GNNs to molecular structures, we must transform the molecule into a numerical representation that can be understood by the model. It is a rather complicated step and it will vary depending on the specific architecture of the GNN model. Fortunately, most of that preprocessing is covered by external libraries such as Deepchem or RDKit.
要將GNN應用于分子結構,我們必須將分子轉換為模型可以理解的數字表示形式。 這是一個相當復雜的步驟,并且會根據GNN模型的特定架構而有所不同。 幸運的是,大多數預處理都被Deepchem或RDKit之類的外部庫所覆蓋。
Here, I will quickly explain the most common approaches to preprocess a molecular structure.
在這里,我將快速解釋預處理分子結構的最常用方法。
微笑 (SMILES)
SMILES is a string representation of the 2D structure of the molecule. It maps any molecule to a special string that is (usually) unique and can be mapped back to the 2D structure. Sometimes, different molecules can be mapped to the same SMILES string which might decrease the performance of the model.
SMILES是分子的2D結構的字符串表示。 它將任何分子映射到(通常)唯一且可以映射回2D結構的特殊字符串。 有時,不同的分子可以映射到相同的SMILES字符串,這可能會降低模型的性能。
指紋識別 (Fingerprints)

Fingerprints is a binary vector where each bit represents whether a certain substructure of the molecule is present or not. It is usually quite long and might fail to incorporate some structural information such as chirality.
指紋是一個二進制向量,其中每個位代表是否存在該分子的某個子結構 。 它通常很長,可能無法合并一些結構信息,例如手性 。
鄰接矩陣和特征向量 (Adjacency Matrix and Feature Vectors)
Another way to preprocess a molecular structure is to create an adjacency matrix. The adjacency matrix contains information about the connectivity of atoms, where “1” means that there is a connection between them and “0” that there is none. The adjacency matrix is sparse and is often quite big which might not be very efficient to work with.
預處理分子結構的另一種方法是創建鄰接矩陣 。 鄰接矩陣包含有關原子連接性的信息,其中“ 1”表示原子之間存在連接,而“ 0”表示不存在連接。 鄰接矩陣是稀疏的,并且通常很大,使用它可能不是很有效。
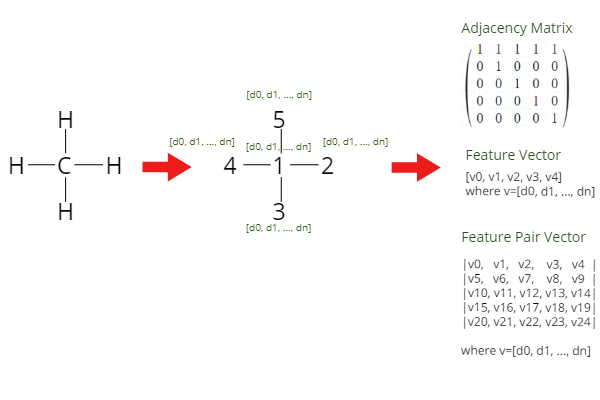
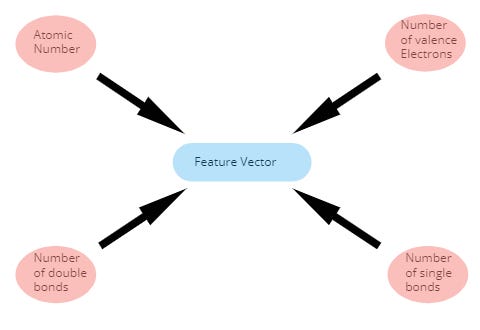
Together with this matrix, we can provide to the GNN model information about each individual atom and information about neighbouring atoms in a form of a vector. In the feature vector for each atom, there can be information about the atomic number, number of valence electrons, or number of single bonds. There is of course many more and they can fortunately be generated by RDKit and Deepchem,
與此矩陣一起,我們可以以矢量的形式向GNN模型提供有關每個單個原子的信息以及有關相鄰原子的信息。 在每個原子的特征向量中,可以包含有關原子序數,價電子數或單鍵數的信息。 當然還有更多,它們可以由RDKit和Deepchem生成,
溶解度 (Solubility)
The variable that we are going to predict is called cLogP and is also known as octanol-water partition coefficient. Basically, the lower is the value the more soluble it is in water. clogP is a log ratio so the values range from -3 to 7 [6].
我們將要預測的變量稱為cLogP和 也稱為辛醇-水分配系數。 基本上,該值越低,它在水中的溶解度越高。 clogP是對數比率,因此值的范圍是-3到7 [6]。
There is also a more general equation describing the solubility logS:
還有一個更通用的方程式描述溶解度logS :
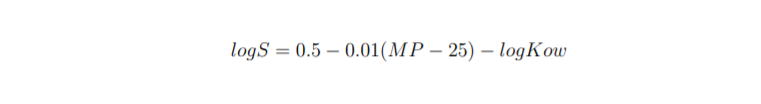
The problem with that equation is that MP is very difficult to predict from the chemical structure of the molecule [7]. All available solubility datasets contain only cLogP value and this is the value that we are going to predict as well.
該方程式的問題在于, 很難通過分子的化學結構來預測MP [7]。 所有可用的溶解度數據集僅包含cLogP值,這也是我們將要預測的值。
Deepchem的動手部分 (Hands-on Part with Deepchem)
Colab notebook that you can run by yourself is here.
您可以自己運行的Colab筆記本在這里。
Deepchem is a deep learning library for life sciences that is built upon few packages such as Tensorflow, Numpy, or RDKit. For molecular data, it provides convenient functionality such as data loaders, data splitters, featurizers, metrics, or GNN models. From my experience, it is quite troublesome to setup so I would recommend running it on the Colab notebook that I’ve provided. Let’s get started!
Deepchem是用于生命科學的深度學習庫,它建立在Tensorflow,Numpy或RDKit等少數軟件包的基礎上。 對于分子數據,它提供了方便的功能,例如數據加載器,數據拆分器,特征化器,度量或GNN模型。 根據我的經驗,設置起來很麻煩,所以我建議在我提供的Colab筆記本上運行它。 讓我們開始吧!
Firstly, we will download a Delaney dataset, which is considered as a benchmark for solubility prediction task. We then load the dataset using CSVLoader class and specify a column with cLogP data which is passed into tasks argument. In smiles_field, name of the column with SMILES string have to be specified. We choose a ConvMolFeaturizer which will create input features in a format required by the GNN model that we are going to use.
首先,我們將下載Delaney數據集,該數據集被視為溶解度預測任務的基準。 然后,我們使用CSVLoader類加載數據集,并指定包含cLogP數據的列,該列將傳遞到task參數。 在smiles_field中,必須指定帶有SMILES字符串的列的名稱。 我們選擇一個ConvMolFeaturizer,它將以我們將要使用的GNN模型所需的格式創建輸入要素。
# Getting the delaney dataset
!wget https://raw.githubusercontent.com/deepchem/deepchem/master/datasets/delaney-processed.csv
from deepchem.utils.save import load_from_disk
dataset_file= "delaney-processed.csv"# Loading the data from the CSV file
loader = deepchem.data.CSVLoader(tasks=["ESOL predicted log solubility in mols per litre"], smiles_field="smiles", featurizer=deepchem.feat.ConvMolFeaturizer())
# Featurizing the dataset with ConvMolFeaturizer
dataset = loader.featurize(dataset_file)Later, we split the dataset using RandomSplitter and divide data into training and validation set. We also use a normalization for y values so they have zero mean and unit standard deviation.
之后,我們使用RandomSplitter分割數據集,并將數據分為訓練和驗證集。 我們還對y值使用歸一化,因此它們的均值和單位標準差為零。
# Splitter splits the dataset # In this case it's is an equivalent of train_test_split from sklearnsplitter = deepchem.splits.RandomSplitter()# frac_test is 0.01 because we only use a train and valid as an exampletrain, valid, _ = splitter.train_valid_test_split(dataset,frac_train=0.7,frac_valid=0.29,frac_test=0.01)# Normalizer will normalize y values in the datasetnormalizer = deepchem.trans.NormalizationTransformer(transform_y=True, dataset=train, move_mean=True)train = normalizer.transform(train)test = normalizer.transform(valid)In this example, we will use a GraphConvModel as our GNN models. It’s an architecture that was created by Duvenaud, et al. You can find their paper here. There are other GNN models as a part of the Deepchem package such as WeaveModel, or DAGModel. You can find a full list of the models with required featurizers here.
在此示例中,我們將使用GraphConvModel作為我們的GNN模型。 這是Duvenaud等人創建的架構。 您可以在這里找到他們的論文。 Deepchem軟件包中還包含其他GNN模型,例如WeaveModel或DAGModel。 您可以在此處找到具有所需功能的所有型號的完整列表。
In this code snippet, a person R2 score is also defined. Simply speaking, the closer this value is to 1, the better is the model.
在此代碼段中,還定義了人員R2分數。 簡單地說,該值越接近1,模型越好。
# GraphConvModel is a GNN model based on
# Duvenaud, David K., et al. "Convolutional networks on graphs for
# learning molecular fingerprints."
from deepchem.models import GraphConvModel
graph_conv = GraphConvModel(1,batch_size=50,mode="regression")
# Defining metric. Closer to 1 is better
metric = deepchem.metrics.Metric(deepchem.metrics.pearson_r2_score)Deepchem models use Keras API. The graph_conv model is trained with the fit() function. Here you can also specify the number of epochs. We get the scores with evaluate() function. Normalizer has to be passed here because y values need to be mapped again to the previous range before computing the metric score.
Deepchem模型使用Keras API。 graph_conv模型是使用fit()函數訓練的。 在這里,您還可以指定時期數。 我們使用評價()函數獲得分數。 必須在此處傳遞規范化器,因為在計算指標得分之前, y值需要再次映射到先前的范圍。
# Fitting the model
graph_conv.fit(train, nb_epoch=10)# Reversing the transformation and getting the metric scores on 2 datasets
train_scores = graph_conv.evaluate(train, [metric], [normalizer])
valid_scores = graph_conv.evaluate(valid, [metric], [normalizer])And that’s all! You can do much more interesting stuff with Deepchem. They created some tutorials to show what else you can do with it. I highly suggest looking over it. You can find them here.
就這樣! 您可以使用Deepchem做更多有趣的事情。 他們創建了一些教程來展示您還可以做什么。 我強烈建議您仔細檢查一下。 您可以在這里找到它們。
Thank you for reading the article, I hope it was useful for you!
感謝您閱讀本文,希望對您有所幫助!
關于我 (About Me)
I am an MSc Artificial Intelligence student at the University of Amsterdam. In my spare time, you can find me fiddling with data or debugging my deep learning model (I swear it worked!). I also like hiking :)
我是阿姆斯特丹大學的人工智能碩士研究生。 在業余時間,您會發現我不喜歡數據或調試我的深度學習模型(我發誓它能工作!)。 我也喜歡遠足:)
Here are my social media profiles, if you want to stay in touch with my latest articles and other useful content:
如果您想與我的最新文章和其他有用內容保持聯系,這是我的社交媒體個人資料:
Medium
中
Linkedin
領英
Github
Github
Personal Website
個人網站
翻譯自: https://towardsdatascience.com/drug-discovery-with-graph-neural-networks-part-1-1011713185eb
吳恩達神經網絡1-2-2
本文來自互聯網用戶投稿,該文觀點僅代表作者本人,不代表本站立場。本站僅提供信息存儲空間服務,不擁有所有權,不承擔相關法律責任。 如若轉載,請注明出處:http://www.pswp.cn/news/391083.shtml 繁體地址,請注明出處:http://hk.pswp.cn/news/391083.shtml 英文地址,請注明出處:http://en.pswp.cn/news/391083.shtml
如若內容造成侵權/違法違規/事實不符,請聯系多彩編程網進行投訴反饋email:809451989@qq.com,一經查實,立即刪除!相關文章

再利用Chakra引擎繞過CFG

SYN flood 攻擊)
重學TCP協議(10)SYN flood 攻擊

大數據入門課程_我根據數千個數據點對互聯網上的每門數據科學入門課程進行了排名...

python 數據框缺失值_Python:處理數據框中的缺失值
 - 推薦)
Spring Cloud 5分鐘搭建教程(附上一個分布式日志系統項目作為參考) - 推薦
)
51nod1832(二叉樹/高精度模板+dfs)
TFO(Tcp Fast Open))
重學TCP協議(11)TFO(Tcp Fast Open)
![[網絡安全] 遠程登錄](http://pic.xiahunao.cn/[網絡安全] 遠程登錄)
[網絡安全] 遠程登錄

外星人圖像和外星人太空船_衛星圖像:來自太空的見解

chrome恐龍游戲_如何玩沒有互聯網的Google Chrome恐龍游戲-在線和離線

Hotpatch潛在的安全風險

spring中@Inject和@Autowired的區別?分別在什么條件下使用呢?

Objective-C語言的動態性

內存泄漏和內存溢出的區別

怎么注銷筆記本icloud_如何在筆記本電腦或臺式機的Web瀏覽器中在線查看Apple iCloud照片

棒棒糖 宏_棒棒糖圖表

ubuntu上如何安裝tomcat
)
leetcode 1734. 解碼異或后的排列(位運算)
