今天給同學們分享一篇實驗文章“Antisense oligonucleotide therapy for H3.3K27M diffuse midline glioma”,這篇文章發表在Sci Transl Med期刊上,影響因子為17.1。

結果解讀:
CRISPR-Cas9消耗H3.3K27M恢復了H3K27三甲基化,并延緩了患者來源的神經球和原位異種移植物的生長
作為對本研究中使用的模型中完全基因敲除H3-3A的生物學后果的控制,作者首先使用CRISPR-Cas9和sgRNA靶向突變和野生型等位基因,但不影響編碼相同蛋白質的H3-3B,在兩個DIPG患者細胞系SU-DIPG-XIII和XVII中(9)。K27M突變對H3K27me3總量的顯性負效應已有充分的文獻證明(1-4)。在攜帶H3-3A雜合突變的兩個患者細胞系中,H3.3K27M敲除恢復了抑制性的H3K27me3三甲基化標記,并減少了通過免疫印跡檢測到的允許性的H3K27ac乙酰化標記。轉導gRNA抗性H3-3A cDNA降低了恢復的H3K27me3標記,與靶向效應一致。敲除恢復了H2K27me3三甲基化并減少了細胞增殖,通過免疫熒光、EdU染色以及軟瓊脂集落形成進行檢測。為了確定H3.3K27M在體內需要用于腫瘤維持,作者在免疫缺陷的非肥胖糖尿病、嚴重聯合免疫缺陷病、伽瑪(NSG)小鼠的出生后第3天(P3)進行了患者細胞或H3-3A K27M 基因敲除細胞的原位移植(9)。作者選擇了SU-DIPG-XIII系列進行此實驗,因為其較慢的增殖速率擴大了作者治療的窗口期。移植的敲除細胞顯示出延遲的腫瘤潛伏期,導致顯著增加的生存率(P = 0.039)。與正常相鄰組織相比,H3.3K27M異種移植瘤在組織學上與患者腫瘤相似,H3K27me3標記全局減少。這種效應在敲除瘤中得到緩解,并與成熟神經元標記物神經核抗原(NeuN)和星形膠質纖維酸性蛋白(GFAP)的表達升高相關,通過免疫熒光檢測到,這表明敲除細胞在體內增殖較少且更傾向于分化的譜系。這些結果證實并擴展了先前的研究(5, 6),并強調了突變H3的潛力。3K27M作為治療靶點。
Gapmer ASO篩選以減少突變的H3-3A mRNA和H3.3K27M蛋白質
為了在DIPG患者細胞中藥理學地靶向H3-3A mRNA,作者探索了ASOs的潛力。作者設計并測試了帶有2'-O-甲氧基乙基(MOE)翼和磷酸硫酸酯(PS)骨架的gapmer ASOs。這些gapmer ASOs具有類似DNA的中央區域,通過內源性RNase H引導對互補mRNA或前mRNA靶點的剪切,并具有化學修飾的翼,促進更緊密的RNA結合,增強穩定性和改善細胞攝取(12)。首先,為了嘗試實現靶向等位基因特異性沉默,作者設計了10個交集的20-mer PS-MOE-ASOs(#1-10),跨越H3-3A外顯子2的突變位點和相鄰核苷酸,突變位點位于ASO間隙上。這些序列與H3-3A K27M 等位基因完全互補,并與H3-3A WT 等位基因有一個錯配。其次,為了實現靶向基因特異性沉默-考慮到野生型H3.3蛋白仍然由H3-3B基因表達-作者設計了另外五個交集的20-mer PS-MOE gapmer ASOs(#11-15),以靶向外顯子2突變位點上游或下游的外顯子區域。這些ASO僅設計用于靶向H3-3A轉錄本,而不是H3-3B轉錄本,或者編碼H3.1或H3.2經典組蛋白蛋白質的其他基因轉錄本。此外,作者通過克隆包含外顯子1至3、完整內含子1和2的基因組片段,生成了一個野生型H3-3A迷你基因和一個帶有A到T突變的突變型H3-3A迷你基因。
為了測試這些ASOs是否介導RNase-H1對H3-3A K27M mRNA的剪切,或者對突變型和野生型H3-3A都起作用,作者將單個ASOs(100 nM)與H3-3A WT 或H3-3A K27M 小基因共轉染到HeLa細胞中。作者還通過自由攝取(4 μM)將這些ASOs傳遞到患者來源的神經球培養中。后一種方法依賴于受體介導的內吞作用,并且需要更高濃度的ASO(12)。作者的初步小基因篩選確定了三個連續的具有等位基因特異設計的ASOs(ASO4、5和6),以及兩個連續的具有基因特異設計的ASOs(ASO12和13),它們實現了H3-3A的強效沉默(圖1A)。所有等位基因特異的ASOs在患者來源的細胞中對突變型等位基因的沉默效果更強,而在這個濃度下,ASO4、5和6在小基因環境中都能強效沉默兩個等位基因。同樣,通過自由攝取傳遞到神經球培養中的ASOs將內源性突變等位基因的表達降低了50-70%,而野生型等位基因的表達降低程度較小(30-40%)(圖1B)。出乎意料的是,一種基因特異性的ASO,ASO15,也促進了等位基因特異性的沉默,這表明了在其靶區域內結合的可能的調控性RNA結合蛋白的立體阻斷(圖1B)。
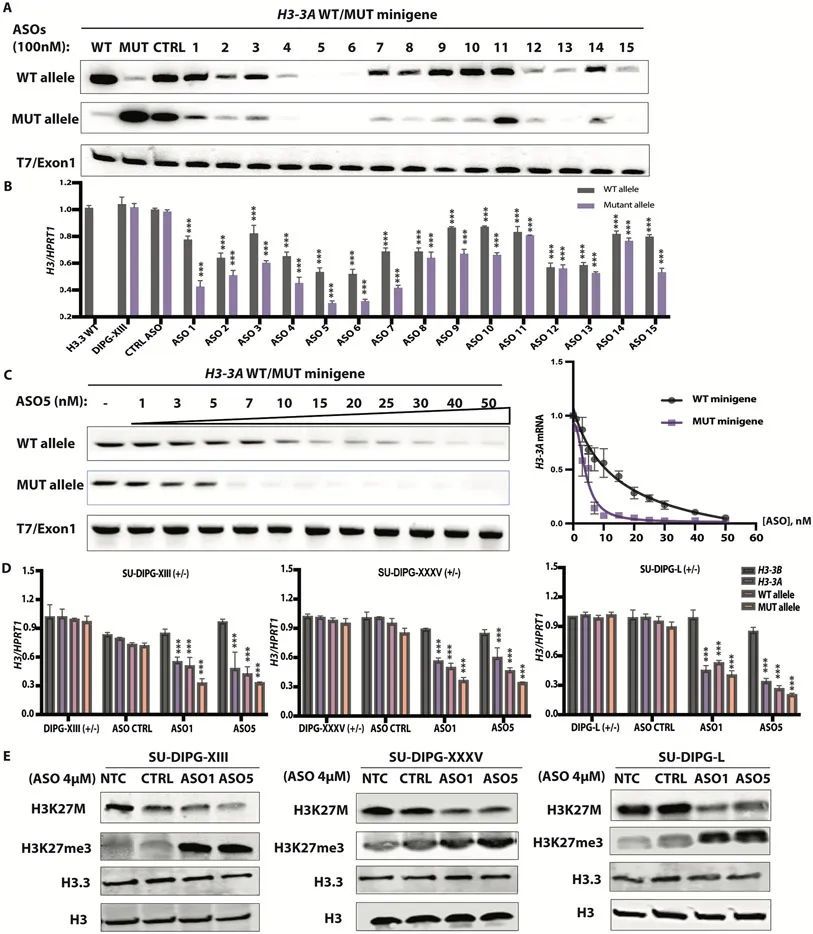
ASO介導的H3.3K27M消減延遲了神經球的生長并改變了細胞形態。
使用多個H3.3K27M DIPG患者衍生的細胞系作為神經球培養,作者觀察到與亂序對照ASO相比,主導的ASO(ASO1和5)明顯延緩了腫瘤細胞的生長。作者觀察到,使用4μM ASO1或ASO5處理的SU-DIPG-XIII、SU-DIPG-XXXV和SU-DIPG-L神經球的增殖明顯減慢(P < 0.001)(圖2A、B和C)。相比之下,對于對照的H3.3WT膠質瘤細胞系,亂序對照ASO和ASO5對增殖沒有可測量的影響。在第5天時點,這三個DIPG患者衍生的細胞系的細胞形態發生了變化,觀察到了類似神經突起的過程(圖2D)。這種形態學變化與作者在CRISPR敲除正位移植模型中觀察到的分化表型一致。此外,使用主導的ASOs處理的SU-DIPG-XIII和SU-DIPG-L細胞系形成了較小的神經球(圖2E)。
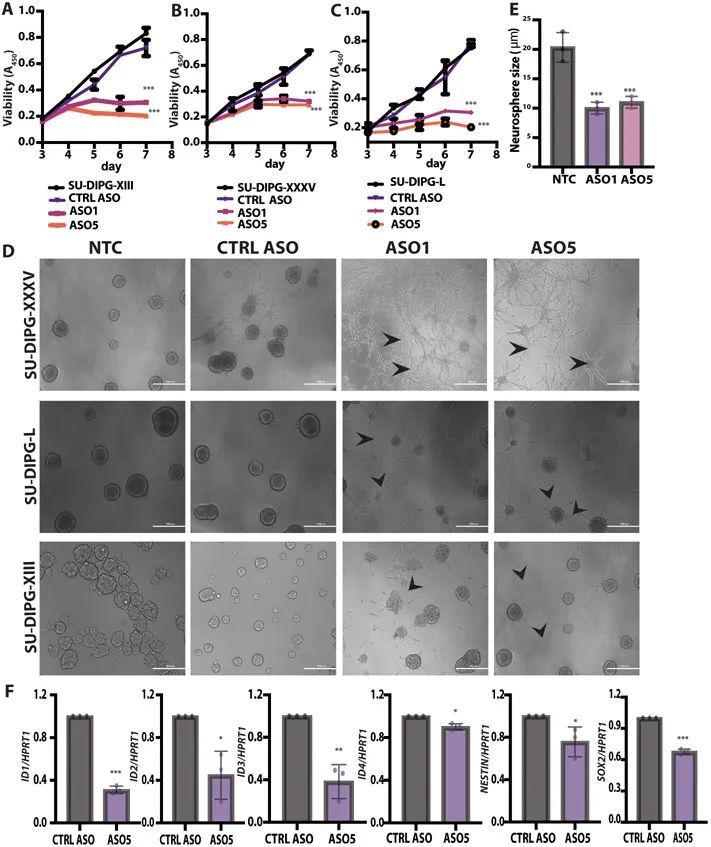
ICV注射鉛ASO促進了H3.3K27M的消耗,在RCAS-Tva小鼠模型中降低了腫瘤級別和促進了分化
為了在體內測試作者的引導ASOs,作者首先采用了RCAS-Tva系統建立了小鼠DIPG模型。RCAS,即具有剪接受體的復制能力的鳥肉瘤-白血病病毒長末端重復序列(LTR),是一種只感染表達鳥Tva逆轉錄病毒受體的細胞的病毒載體(13)。此系統先前被用于顯示小鼠組蛋白H3.3K27M或H3.1K27M加速膠質瘤發生,與另一個遺傳小鼠模型的結果一致(14-16)。作者通過引入人H3-3A cDNA來改進該系統,其轉錄物可以被作者的人類特異性ASOs靶向。作者將產生編碼P1噬菌體Cre重組酶、人H3.3K27M、小鼠血小板源性生長因子B(PDGFB)和螢火蟲熒光素的雞DF1細胞輸送到由小鼠Nestin啟動子驅動的帶有Tva和轉化相關蛋白53(Trp53)-floxed等位基因的新生小鼠的腦干中(圖3A)。小鼠在3-6周內發展出高級別的膠質瘤,局部位于中線區域。通過組織學分析,作者發現FLAG標記的H3.3K27M存在于膠質瘤病變中。大約90%的H3.3K27M腫瘤顯示全局H3K27me3減少,與正常相鄰組織相比,呈現出與患者腫瘤組織學相似的模式。腫瘤細胞也呈陽性染色表達寡突膠質細胞譜系標記物寡突膠質細胞轉錄因子2(OLIG2)(15)。
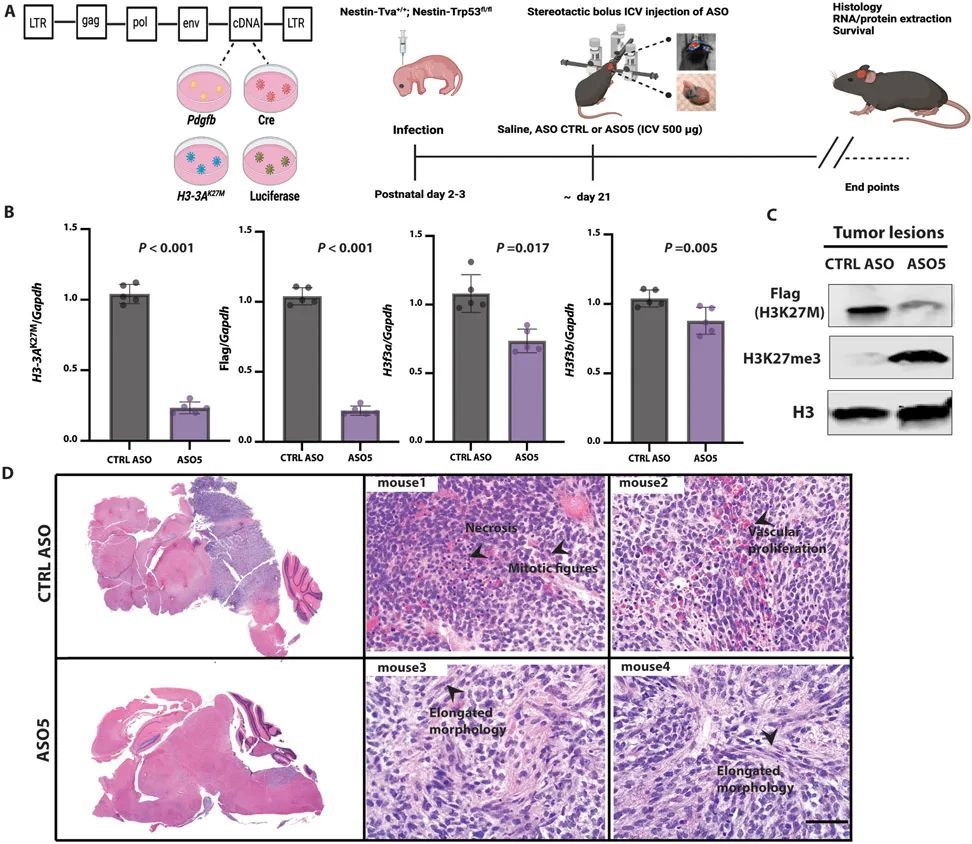
為了測試體內引物ASO的有效性,作者進行了立體定向ICV注射,將其直接輸送到腦脊液中(CSF)(17, 18)。作者在腫瘤發生時,通過生物發光成像(圖3A)檢測到的時間點,向側腦室注射了一次性劑量(500 μg)的引物ASO5或CTRL ASO溶于鹽水中。作者在預設的時間點從腫瘤或正常相鄰組織中提取RNA和蛋白質。ASO5顯著(P < 0.001)降低了人類H3-3A K27M mRNA和FLAG標簽的表達,對內源性小鼠野生型H3f3a的影響較小,但對H3f3b沒有影響(圖3B)。在蛋白質水平上,ASO5顯著(P < 0.001)降低了人類FLAG標記的H3.3K27M,并增加了H3K27me3的豐度(圖3C)。作者進一步觀察到FLAG標記的免疫熒光染色的消失,即在約80%的處理細胞中,FLAG信號低于檢測水平。
接受對照ASO處理的小鼠發展出高度增殖和侵襲性的膠質瘤,伴有大量有絲分裂圖像、廣泛的血管增生、偶爾的壞死以及壞死區周圍的偽假性帕拉賽德。這些腫瘤沒有包膜,邊界不清晰,在腫瘤-腦界面處有一些腫瘤侵襲。相比之下,接受ASO5處理的小鼠顯示出腫瘤生長的潛伏期延長,腫瘤呈現出延長的形態(圖3D)。在這些腫瘤病變中,有絲分裂圖像很少,壞死并不明顯。此外,ASO5處理的腫瘤病變中的細胞形態學上類似于膠質細胞和成熟神經元。作者得出結論,體內由引導ASO引起的突變H3-3A的沉默導致了低級別腫瘤形成和更多分化的外觀。
為了進一步描述組織學觀察到的表型,作者對已知分化標記物進行了免疫熒光染色。在具有相同遺傳背景但攜帶H3.3WT腫瘤的小鼠中,作者檢測到成熟星形膠質細胞(GFAP)(19)、神經元(NeuN)(20)和少突膠質細胞(髓鞘堿性蛋白(MBP))(21)的標記物,表明H3.3WT膠質瘤中發生了神經發生和膠質發生(圖4A)。相比之下,在存在H3.3K27M的腫瘤病灶中,檢測到的GFAP、NeuN和MBP細胞較少(圖4B,頂部面板)。經過一次腦室內注射ASO5劑量后,作者檢測到大量的GFAP、NeuN和MBP細胞,與Ki67標記的增殖細胞數量減少相一致,Ki67是一個在細胞周期的所有活躍階段表達的核細胞增殖相關抗原(圖4B,底部面板)。Ki67的減少和GFAP、NeuN標記的增加在統計學上具有顯著性(P = 0.002或0.003)(圖4C和D)。作者通過對從正常相鄰組織和腫瘤病灶中提取的總蛋白進行免疫印跡驗證了這些結果(圖4E)。作者得出結論,H3.33K27M阻礙了星形膠質細胞和神經元的分化,而ASO介導的H3.3K27M消耗恢復了分化程序。

ICV注射ASO5降低了NESTIN細胞群體,并延長了腫瘤生長的潛伏期
DMG在特定的時空背景下出現,并在中童期發生,發病高峰年齡為6至9歲。細胞起源和微環境對腫瘤生長至關重要(26, 27)。先前的回顧性克隆分析顯示,NESTIN + 細胞在人類中腦、腦橋和延髓中富集,貫穿整個童年期,且在腹側腦橋處密度最高;因此,NESTIN + 細胞群體與DIPG的時空發病率完全一致(27)。在作者的小鼠模型中,相對于正常相鄰腦組織,NESTIN + 細胞在腫瘤病變部位富集。經過ASO5治療后,NESTIN + 細胞群體明顯減少,與GFAP + 細胞數量的增加呈負相關(圖4F)。此外,ASO5治療的小鼠的存活時間顯著延長(P < 0.0001),與對照組ASO治療的小鼠相比(圖4G)。這些結果表明,H3.3K27M腫瘤起源于神經干細胞(NESTIN + ),并與TRP53缺失和PDGF信號傳導相關。
ASO5治療在RCAS-Tva模型中產生的分化細胞是腫瘤起源的。
為了確定ASO5處理后出現的分化細胞是否來自腫瘤,而不是小鼠細胞被招募到損傷部位,作者利用在RCAS-Tva模型中表達的血凝素(HA)標記的Pdgfb和Cre cDNAs。作者使用HA抗體進行免疫熒光染色,并觀察到大部分腫瘤細胞都是HA + 。當作者用HA和各個分化標記物雙標記細胞時,作者觀察到大量的HA + 星形膠質細胞、神經元和少突膠質細胞(圖5A)。
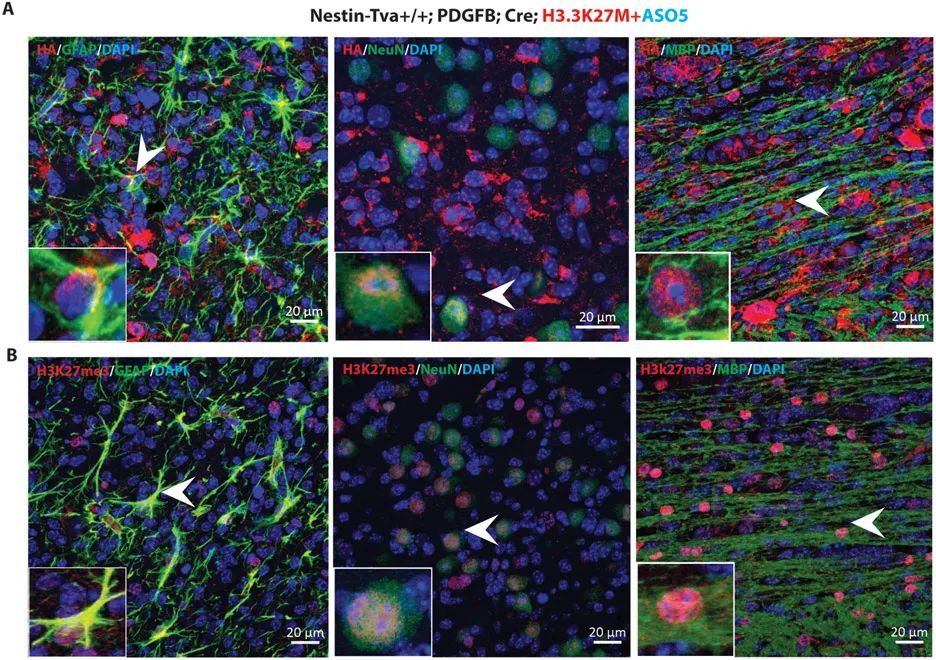
ICV注射ASO5促進了星形膠質細胞、神經元和少突膠質細胞的分化,并減少了DIPG患者源自移植模型的腫瘤增殖
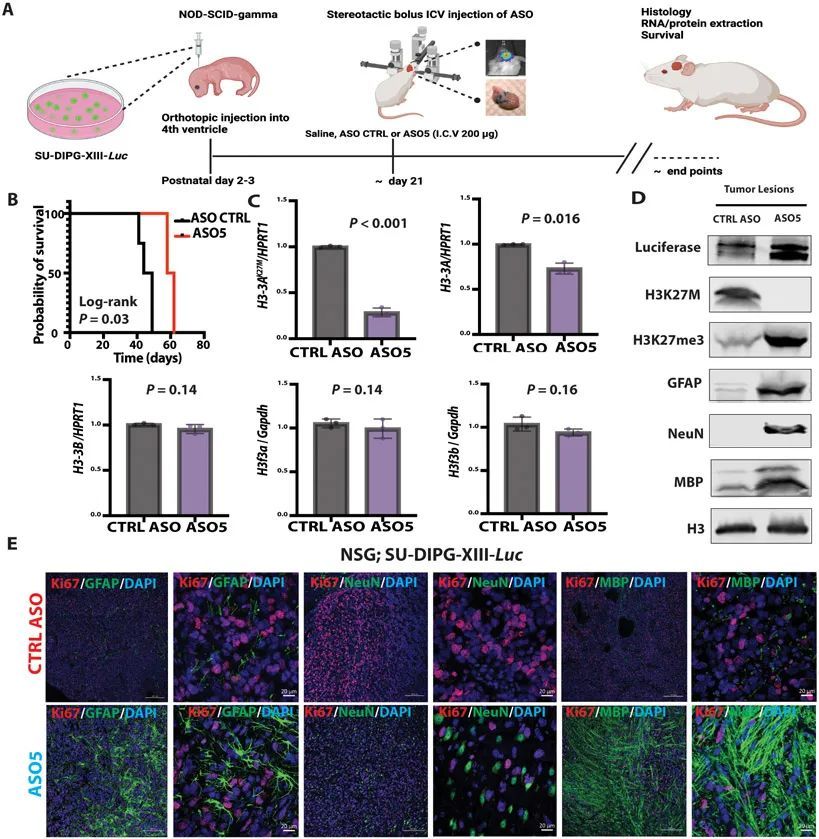
為了確認和擴展上述觀察結果,作者還在一個原位異種移植小鼠模型中測試了ASO5,如(9)所述,使用圖3中顯示的SU-DIPG患者系列之一。作者將10個表達熒光素的SU-DIPG-XIII細胞注射到P3免疫缺陷的NSG小鼠的第4腦室中,并使用生物發光成像來跟蹤腫瘤發生。由于這些免疫缺陷小鼠無法耐受RCAS-Tva模型中使用的高濃度ASO,作者對其進行了單次腦室注射,注射200μg對照ASO或ASO5(圖6A)。與對照ASO處理組相比,ASO5處理組的小鼠存活時間顯著延長(P = 0.03)(圖6B)。與RCAS-Tva小鼠模型類似,ASO5顯著(P < 0.001;P = 0.016)降低了人類H3-3A mRNA和總人類H3-3A mRNA的表達,但不影響內源性小鼠野生型H3f3a或H3f3b(圖6C)。此外,ASO5降低了人類H3.3K27M蛋白的表達,并增加了H3K27me3修飾的程度。ASO5處理還導致Luciferase標記的SU-DIPG-XIII細胞系中GFAP、NeuN和MBP蛋白的豐度升高(圖6D)。ASO5處理的小鼠在腫瘤病灶中表現出不同的表型(GFAP + ,NeuN + ,和MBP + ),細胞增殖較少(Ki67 + ),而在對照組ASO處理的小鼠中,神經發生和膠質發生受到損害,大部分腫瘤細胞仍處于增殖狀態(圖6E)。與RCAS-Tva模型類似,表達各種分化標記的細胞也具有H3K27me3 + 。
總結
總之,作者開發了在體外和體內直接靶向癌組蛋白mRNA的gappmer ASOs。前導ASO有效降解H3-3AK27M mRNA,減少患者來源細胞和小鼠模型中的H3.3K27M蛋白。H3.3K27M蛋白豐度的降低導致小鼠模型中神經發生和膠質發生的恢復,腫瘤生長的潛伏期延長,存活率增加。藥物干預的效果不如預期的完全基因敲除,因為ASO治療減少但不會消除突變蛋白的表達。此外,與植入前基因敲除不同,治療性ASO是在腫瘤發作后給藥的,這代表了一種更現實的臨床情況。作者預計,最大的臨床療效可能需要聯合治療,包括ASO給藥,例如放療(36)或CAR-T細胞免疫療法(37,38)。



















-源碼)