在前面兩篇文章中,我們已經成功搭建了R和RStudio這一強大的生信分析平臺。然而,工具再好,若不懂得如何“放置”和“理解”你的數據,一切都將寸步難行。今天,我們將學習R語言最重要的部分——數據類型(Data Types)?和?數據結構(Data Structures)。
測序數據一般包括基因名、基因ID、表達量、樣本信息、分組標簽……如果這些數據雜亂無章,你如何能從中挖掘出有意義的生物學結論?R語言通過其精妙的數據類型和豐富的數據結構,為我們提供了將“數據亂碼”整理成“有序信息”的強大能力。
本篇,我們不僅僅停留在概念講解,而是將這些基礎知識與真實的生信分析場景緊密結合,在解決問題的過程中,體會數據管理的藝術。
R語言的數據類型:數據的“本質屬性”
在R中,每個數據都有其特定的數據類型,這決定了R如何存儲和處理它。理解數據類型是后續構建數據結構的基礎。最常見的幾種數據類型包括:
- 數值型(Numeric):用于存儲數字,包括整數(Integer)和雙精度浮點數(Double)。
應用場景: 基因表達量(如TPM、FPKM、Counts)、樣本年齡、實驗重復次數等。
示例:?c(100.5, 230, 0.01) - 字符型(Character):用于存儲文本信息。
應用場景: 基因ID(如"TP53")、樣本名稱(如"Sample_WT_Rep1")、通路名稱、GO富集結果的描述等。
示例:?c("GeneA", "GeneB", "GeneC") - 邏輯型(Logical):只包含 TRUE (真) 或 FALSE (假) 兩種值,常用于條件判斷。
應用場景: 判斷基因是否上調/下調、樣本是否屬于疾病組、質控是否通過等。
示例:?c(TRUE, FALSE, TRUE) - 因子型(Factor):特殊的一種字符型,用于存儲有限個預定義類別的數據,在統計建模中非常重要。R會將其視為分類變量。
應用場景: 實驗分組(如“Control” vs “Treatment”)、性別(“Male” vs “Female”)、細胞類型(“B_cell”, “T_cell”, “Macrophage”)等。
示例:?factor(c("Control", "Treatment", "Control"))
為何重要: 在進行差異表達分析或回歸分析時,R會自動識別因子變量,并將其正確地納入統計模型,處理起來比普通字符型更加高效和準確。
R語言的數據結構:數據的“組織形式”
數據類型給數據“安上了標簽”,數據結構就是將這些帶標簽的數據梳理成有規律的數據集,便于函數處理,進而發現生物學規律。
向量(Vector):一維數據的序列
定義: 向量是R中最基本的數據結構,用于存儲同一種數據類型的元素序列。
應用場景:
單個基因在不同樣本中的表達量。
一系列樣本的測序深度。
一組基因的差異表達P值。
單個樣本的質控指標(如線粒體基因比例)。
操作與理解:
使用?c()?函數創建向量。
通過索引([])訪問和修改元素。
進行向量化的運算(無需循環,R會自動對每個元素執行操作,效率高)。
【實戰演練:批量計算P值校正】
假設我們通過差異表達分析得到了10個基因的原始P值,現在需要進行FDR(False Discovery Rate)校正。
# 1. 創建一個數值型向量,存儲原始P值
raw_p_values <- c(0.001, 0.045, 0.12, 0.0005, 0.08, 0.003, 0.5, 0.06, 0.009, 0.02)
print("原始P值:")
print(raw_p_values)# 2. 使用p.adjust函數進行FDR校正(這是一個典型的向量化操作)
# "fdr" 是校正方法,也可以是 "bonferroni", "BH" 等
adjusted_p_values <- p.adjust(raw_p_values, method = "fdr")
print("FDR校正后的P值:")
print(adjusted_p_values)# 思考:如果raw_p_values中混入了字符型數據,例如 "NA",p.adjust函數還能正常工作嗎?為什么?

矩陣(Matrix):二維的同類型數據表
定義: 矩陣是二維的數組,所有元素必須是同一種數據類型。它有行和列,非常適合存儲表格型數據。
應用場景:
基因表達矩陣: 行是基因,列是樣本,矩陣中的值是基因的表達量(數值型)。這是轉錄組和單細胞分析中最核心的數據結構之一。
距離矩陣、相似性矩陣等。
操作與理解:
使用?matrix()?函數創建。
通過?dim()?查看維度,nrow()?獲取行數,ncol()?獲取列數。
通過?matrix[row_index, col_index]?進行索引。
【實戰演練:主成分分析(PCA)的數據準備】
在轉錄組或單細胞分析中,PCA是一種常用的降維和可視化方法,用于查看樣本間的整體關系或細胞群的異質性。PCA通常需要一個基因表達矩陣作為輸入。
場景: 假設我們有3個樣本(SampleA, SampleB, SampleC),和4個基因(Gene1-Gene4)的表達量。
# 1. 創建一個基因表達矩陣
# 數據順序:按列填充,即先填SampleA的Gene1-4,再填SampleB的Gene1-4...
expression_data <- matrix(c(10, 15, 20, 25, # SampleA12, 18, 22, 28, # SampleB30, 25, 18, 12), # SampleCnrow = 4, ncol = 3,byrow = FALSE, # 默認按列填充,即先填滿第一列再第二列dimnames = list(c("Gene1", "Gene2", "Gene3", "Gene4"), # 行名:基因c("SampleA", "SampleB", "SampleC") # 列名:樣本)
)
print("基因表達矩陣:")
print(expression_data)# 2. 對矩陣進行轉置,使行是樣本,列是基因(PCA通常要求樣本在行)
# t() 函數用于轉置矩陣
transposed_expression_data <- t(expression_data)
print("轉置后的矩陣(樣本在行,基因在列):")
print(transposed_expression_data)# 3. 對轉置后的矩陣執行PCA (這里只演示輸入,不深入PCA細節)
# prcomp() 是R中進行PCA的函數
pca_results <- prcomp(transposed_expression_data, scale. = TRUE) # scale. = TRUE 標準化數據
print("PCA結果(部分):")
print(pca_results$x) # 顯示主成分坐標# 思考:為什么PCA需要將矩陣轉置?如果矩陣中存在字符型數據(比如不小心把某個表達量寫成了"low"),prcomp函數能運行嗎?

數據框(Data Frame):最常用的表格型數據結構
定義: 數據框是R中最常用、最靈活的表格型數據結構。它類似于一個電子表格,每列可以是不同的數據類型,但每列內的元素必須是同種類型。每行代表一個觀察值,每列代表一個變量。
應用場景:
差異表達分析結果表: 一列是基因名(字符型),一列是log2FC(數值型),一列是P值(數值型),一列是FDR(數值型),一列是基因ID(字符型)等。
樣本信息表: 樣本ID、分組、年齡、性別等。各種統計分析的輸入數據。
操作與理解:
使用?data.frame()?創建。
通過$符號或[]索引來訪問列。
強大的數據篩選、排序、合并能力。
【實戰演練:繪制火山圖的數據準備與篩選】
火山圖是差異表達分析中最常見的可視化圖表,它結合了基因表達變化的倍數(Log2FC)和統計顯著性(P值),幫助我們快速識別差異表達基因。火山圖的繪制,正是數據框操作的典型應用。
場景: 我們得到了一份包含基因ID、log2FC和FDR值的差異表達分析結果。
# 1. 創建一個數據框,模擬差異表達分析結果
diff_expression_df <- data.frame(GeneID = c("GeneA", "GeneB", "GeneC", "GeneD", "GeneE", "GeneF", "GeneG", "GeneH"),log2FC = c(2.5, -1.8, 0.1, 3.0, -0.5, -2.2, 0.8, 1.5),FDR = c(0.001, 0.008, 0.85, 0.0001, 0.60, 0.002, 0.45, 0.015)
)
print("原始差異表達結果數據框:")
print(diff_expression_df)# 2. 添加一列 -log10(FDR),用于火山圖Y軸,數值越大表示越顯著
diff_expression_df$neglog10FDR <- -log10(diff_expression_df$FDR)
print("添加-log10(FDR)列后的數據框:")
print(diff_expression_df)# 3. 根據閾值篩選差異表達基因
# 篩選標準:log2FC絕對值大于2 且 FDR小于0.05
significant_genes <- subset(diff_expression_df, abs(log2FC) > 2 & FDR < 0.05)
print("篩選出的顯著差異表達基因:")
print(significant_genes)# 4. 準備繪制火山圖 (這里不實際繪制,只展示數據輸入)
# 繪制火山圖的函數(例如ggplot2::ggplot)會直接使用這個數據框的列
# library(ggplot2)
# ggplot(diff_expression_df, aes(x = log2FC, y = neglog10FDR)) +
# geom_point(aes(color = ifelse(abs(log2FC) > 2 & FDR < 0.05, "Significant", "Non-significant"))) +
# geom_vline(xintercept = c(-2, 2), linetype = "dashed", color = "grey") +
# geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "grey") +
# theme_minimal()
# 思考:如果GeneID列是數值型,或者log2FC列是字符型,會發生什么?為什么?

列表(List):最靈活的數據“容器”
定義: 列表是R中最靈活的數據結構,可以存儲任意類型、任意長度的R對象。它可以是向量、矩陣、數據框,甚至可以是另一個列表。
應用場景:
單細胞分析對象: 單細胞分析中,一個?Seurat?對象或?SingleCellExperiment?對象,本質上就是一個大型列表,其中包含:
a. 一個稀疏矩陣(表達量數據)。
b. 一個數據框(細胞的元數據,如細胞類型、批次信息)。
c. 另一個數據框(基因的元數據)。
d. 多個矩陣(降維結果,如PCA、UMAP坐標)。
e. 多個列表(聚類結果、差異基因列表等)。
存儲復雜的統計模型結果。一次性返回多個不同類型的結果。
操作與理解:
使用?list()?函數創建。
通過?[[索引]]?或?$?符號訪問列表中的元素。
【實戰演練:單細胞數據對象的抽象理解】
在單細胞RNA測序(scRNA-seq)分析中,我們通常不會直接操作原始矩陣,而是使用專門的R包(如Seurat或Bioconductor的SingleCellExperiment)構建一個復雜的“單細胞對象”。這個對象本質上就是一個包含多種數據結構的列表。
場景: 抽象模擬一個簡化的單細胞數據對象,其中包含表達矩陣、細胞分組信息和降維坐標。
# 1. 模擬單細胞表達矩陣 (通常是稀疏矩陣,這里用普通矩陣簡化)
# 5個基因,10個細胞
sc_expression_matrix <- matrix(sample(0:10, 50, replace = TRUE), # 隨機生成0-10的整數表達量nrow = 5, ncol = 10,dimnames = list(paste0("Gene", 1:5),paste0("Cell", 1:10))
)# 2. 模擬細胞的元數據(例如細胞類型)
cell_metadata <- data.frame(CellID = paste0("Cell", 1:10),CellType = factor(c(rep("T_cell", 4), rep("B_cell", 3), rep("Macrophage", 3))),Batch = factor(c(rep("Batch1", 5), rep("Batch2", 5)))
)# 3. 模擬降維后的UMAP坐標
umap_coordinates <- matrix(rnorm(20), # 2列,10行,代表10個細胞的2D UMAP坐標nrow = 10, ncol = 2,dimnames = list(paste0("Cell", 1:10), c("UMAP_1", "UMAP_2"))
)# 4. 將這些數據組織成一個列表,模擬單細胞對象
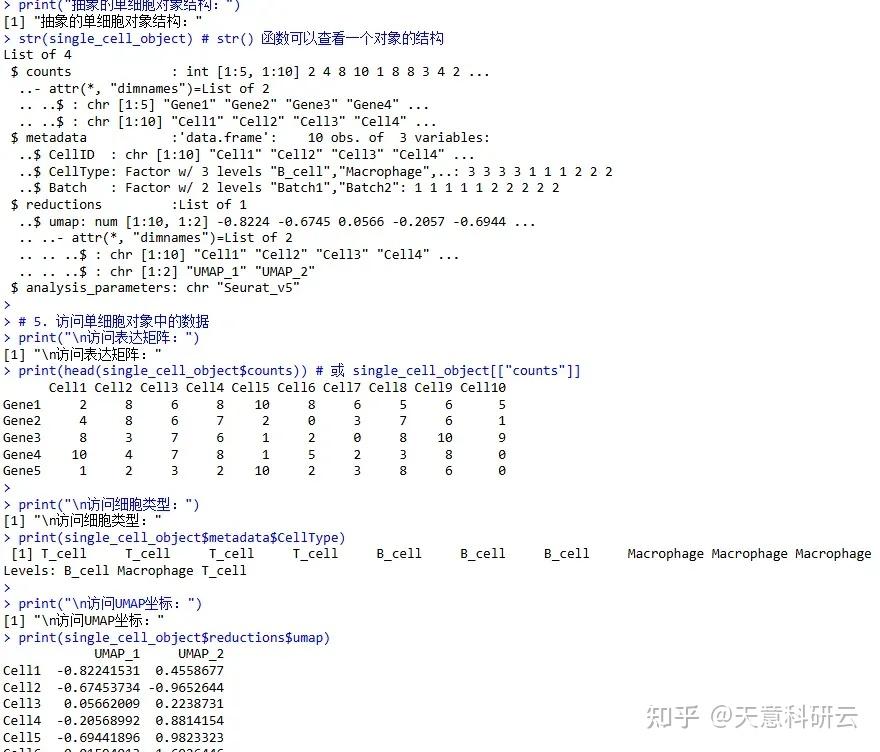
single_cell_object <- list(counts = sc_expression_matrix,metadata = cell_metadata,reductions = list(umap = umap_coordinates),analysis_parameters = "Seurat_v5"# 可以存儲分析版本等額外信息
)print("抽象的單細胞對象結構:")
str(single_cell_object) # str() 函數可以查看一個對象的結構# 5. 訪問單細胞對象中的數據
print("\n訪問表達矩陣:")
print(head(single_cell_object$counts)) # 或 single_cell_object[["counts"]]print("\n訪問細胞類型:")
print(single_cell_object$metadata$CellType)print("\n訪問UMAP坐標:")
print(single_cell_object$reductions$umap)# 思考:為什么單細胞對象要設計成列表這種結構,而不是直接用一個大矩陣或數據框?
# 提示:考慮到不同類型的數據(數值、字符、分類),以及不同維度的數據(基因數x細胞數,細胞數x特征數)。
總結與展望:有序數據,高效分析
通過今天的學習和實踐,你現在應該對R語言的數據類型和數據結構有了更深刻的認識。我們通過轉錄組的差異表達分析和火山圖繪制,以及單細胞數據處理和PCA的數據準備,看到了向量、矩陣、數據框和列表如何在實際生信項目中發揮核心作用。
- 數據類型是數據的屬性,它決定了數據能進行哪些操作。
- 數據結構是數據的組織形式,它決定了數據如何被高效存儲和訪問。
- R Project則為這些有序的數據提供了一個規范的“家”,保證了分析的可重復性。
思考題答案提示:
- 向量P值校正問題: 如果?
raw_p_values?中混入了字符型數據,p.adjust?函數會報錯,因為該函數期望所有輸入都是數值型。R的向量是同質的,如果強制將數值和字符混合,所有元素會被強制轉換為字符型。 - PCA矩陣轉置問題:?
prcomp()?函數默認期望輸入數據的行是觀測值(通常是樣本),列是變量(通常是基因)。因此,如果你的原始矩陣是基因在行、樣本在列,就需要轉置。如果矩陣中存在字符型數據,prcomp()?函數會報錯,因為它只能處理數值型數據。 - 火山圖數據框問題: 如果?
GeneID?列是數值型,雖然可能不會立刻報錯,但在后續如果需要進行基因名匹配等操作時會出錯。如果?log2FC?列是字符型,abs()?函數和?subset()?函數的數值比較會報錯,因為它們期望數值型輸入。 - 單細胞對象列表設計問題: 單細胞對象之所以設計成列表,是因為它需要在一個單一的結構中存儲多種不同類型和不同維度的數據。表達矩陣是數值型二維數據,細胞元數據是包含多種類型的二維數據框,降維坐標是數值型二維數據,而分析參數可能是字符型或數值型。列表的異質性和嵌套性,使其成為容納這些復雜且多樣化數據的理想“容器”。如果使用單一矩陣或數據框,則無法同時滿足所有數據的存儲需求。


![[1Prompt1Story] 注意力機制增強 IPCA | 去噪神經網絡 UNet | U型架構分步去噪](http://pic.xiahunao.cn/[1Prompt1Story] 注意力機制增強 IPCA | 去噪神經網絡 UNet | U型架構分步去噪)
![【P7071 [CSP-J2020] 優秀的拆分 - 洛谷 https://www.luogu.com.cn/problem/P7071】](http://pic.xiahunao.cn/【P7071 [CSP-J2020] 優秀的拆分 - 洛谷 https://www.luogu.com.cn/problem/P7071】)











)



