文獻介紹
?

文獻題目: Hiplot:一個綜合且易于使用的 Web 服務,用于增強出版物準備的生物醫學數據可視化
研究團隊: Openbiox/Hiplot 社區
發表時間: 2022-07-05
發表期刊: Briefings in Bioinformatics
影響因子: 13.99(2022年)
DOI: 10.1093/bib/bbac261
?
摘要
在臨床(clinical)、組學(omics)和機制(mechanism)實驗過程中產生的復雜生物醫學數據,正日益通過基于云計算和可視化的數據挖掘技術得到深入開發。然而,科學界目前仍缺乏一個易于使用的網絡服務平臺,用于生物醫學數據的全面可視化,尤其是能夠根據用戶需求靈活縮放、隨時更新且達到期刊出版級質量的可視化圖表。為此,作者推出一個社區驅動的現代化網絡服務,Hiplot(https://hiplot.org),為生命科學和生物醫學領域提供簡潔優質的數據可視化應用。該平臺使用戶能便捷地通過交互方式完成若干專業化可視化任務,而這些任務以往只能由資深生物信息學或生物統計學研究者完成。該服務配備 240+ 生物醫學數據可視化功能,涵蓋基礎統計、多組學分析、回歸分析、聚類分析、降維分析、薈萃分析、生存分析、風險模型構建等方向,可滿足生物醫學研究者的大部分日常需求。為提升插件使用和開發效率,作者在網站客戶端/服務端引入了多項核心優勢:基于電子表格的數據導入、跨平臺命令行控制器(Hctl)、多用戶管道計算集群、基于 JSON 的插件系統、簡易的數據/參數-結果-錯誤復現機制以及實時更新模式。通過演示數據集/真實數據集和基準測試,作者基于精選原生插件對統計參數、癌癥基因組圖譜、疾病風險因素及網站性能進行了系統評估。平臺訪問量和用戶數量的統計數據進一步印證了該服務對相關領域的潛在影響力。這項新興的免費網絡服務將為生命科學與數據科學領域的研究者提供重要支持。
?
前言
對源自實驗檢測、組學研究和臨床觀察的多維生物醫學數據進行探索與挖掘,主要依賴于現代圖形學與統計學方法,例如疾病診斷和統計描述/推斷。提升假設驅動型與數據驅動型科學研究的可解釋性、可重復性和有效性,正是可視化數據挖掘技術應當發揮的關鍵作用。十余年前,用戶只能通過功能有限、擴展性不足的桌面應用程序進行日常科研數據可視化與分析。而如今,具備更優擴展性的基于云的網絡應用,已成為缺乏編程技能的生物學家和臨床工作者處理復雜生物醫學數據的理想選擇。并且,隨著 Galaxy、DNAnexus 等知名生物信息學云服務的建立,序列比對、變異檢測、表觀遺傳分析等基于流程的上游數據分析任務已得到一定簡化。
然而在這些平臺中,基于表格數據的出版級科學圖表與交互式數據挖掘等典型下游功能仍顯不足。例如知名生物信息云平臺 Galaxy 雖提供數十個可視化插件,但對輕量級生物醫學可視化任務的優化仍不充分;St. Jude 兒童研究醫院云門戶的可視化模塊雖有 20 個基于 JavaScript 的癌癥基因組交互插件,卻缺乏基礎科研圖表功能;而 imageGP 自 2017 年以來僅開發了 16 項科學圖表與分析子功能。要滿足多樣化的可視化需求,仍需科學界通力合作。研究顯示,復雜的用戶界面和薄弱的交互性已成為阻礙用戶采用生物醫學數據可視化工具的主要因素。例如,現有網絡工具鮮少支持在線電子表格的數據預覽與編輯功能,對出版級排版中多圖自動排列(如每頁4/6/9圖)也普遍欠缺考量。此外,任務輸出延遲、缺乏跨平臺命令行工具、數據/參數-結果-錯誤復現困難等問題,進一步限制了網絡工具在生物醫學可視化中的廣泛應用。
值得注意的是,在整體網站設計中需通過核心功能優化網絡服務,以提升效率并降低時間成本。同時,構建全面可擴展的可視化網絡服務面臨的關鍵挑戰在于:如何簡化用戶操作與開發者工作流程。這需要精心設計的前端客戶端/命令行工具、高質量輸出生成、高效完成基礎任務的能力、完善的用戶支持服務以及低門檻的數據/參數-結果-錯誤復現機制。此外,平臺還需通過長期維護持續更新,提升插件的可用性與定制化程度。建立一個專注于出版級生物醫學可視化的開放協作社區,將加速復雜生物醫學網絡應用的構建、驗證與更新。
為應對這些挑戰,作者推出了易于使用和可擴展的網絡服務,Hiplot(https://hiplot.org)與跨學科社區Openbiox(https://openbiox.org),專注于生物醫學數據可視化交互應用的開發。自 2019 年 10 月以來,Hiplot/Openbiox 協作組已開發數百個基于可視化的數據挖掘交互插件。Hiplot 核心功能基于開源方法構建,覆蓋生物學家與臨床工作者最常見的日常可視化需求,并采用簡潔 UIs 與高效交互設計。例如可切換的編輯表格與文件上傳/路徑選擇視圖,顯著提升了數據導入、預覽與導出效率。同時,基于多用戶管道計算集群的響應策略結合版本控制,大幅降低了基礎科研圖表生成的時間成本。而且,平臺還實現了基于 JSON 的結構化插件系統,包含預置 UI 組件、R 開發庫和在線預覽器,以持續擴展功能。通過原生插件的基準測試及訪問量/用戶數統計,作者評估了平臺性能,這些數據將為 Hiplot 及其他同類網 web/R 工具的迭代更新提供重要參考。作者期待這個實時更新的工具包能發展成為生物醫學、生命科學與數據科學領域重要的可視化基礎設施。
?
研究結果
1. Hiplot 的全面功能和優勢概述
自 2019 年以來,Hiplot/Openbiox 聯盟已開發了大量基于網絡的交互式可視化應用(240+),用于生物醫學數據挖掘(Figure 1, Table S1)。另外,通過系統的文獻調研,本研究被證實是為數不多通過社區協作建立的免費網絡服務之一,能夠交互式、全方位地生成出版級生物醫學可視化圖表(Table 1)。觀察顯示,Hiplot 平臺(native Hiplot、R Shiny、Python Streamlit)提供的交互式可視化工具在數量與多樣性上均優于同類平臺。例如,該網絡服務可提供與知名商業科學繪圖軟件 GraphPad 相媲美的現代統計圖表功能(Figure 1)。因此,用戶無需受操作系統或軟件環境限制,即可通過這些開放獲取的可視化功能處理日常數據分析需求,包括數據相關性、分布特征、百分比構成、演化趨勢、流動關系、排序比較及空間特征等維度的展示與統計推斷。
?

Figure 1. Hiplot web 服務中核心功能和優勢的概述
頂部面板展示了網站核心功能分類及其相關受益領域。該平臺已構建了涵蓋現代統計圖表、組學分析和臨床模型的完整生物醫學可視化功能體系,用戶可直接使用 Hiplot 生成符合出版要求的高質量可視化圖表。圖庫展示了基礎圖表、組學數據和臨床模型相關插件的部分應用案例及輸出效果,更多示例可直接在網站應用卡片列表中查看。底部面板突出了提升使用/開發效率和用戶體驗的核心優勢:基于電子表格和可切換文件上傳器的設計簡化了網絡輕量化可視化任務的數據導入流程;依托多用戶管道計算集群可在數秒內完成基礎圖表生成,同時支持分布式部署和 R 運行環境的版本控制;采用基于 JSON 的插件系統及預置UI/后端功能模板,有效減少了輕量化生物醫學可視化任務的冗余開發步驟;網站根據用戶建議實時更新,已解決的個性化需求幫助設置了更多控制圖形輸出的自定義參數。
?
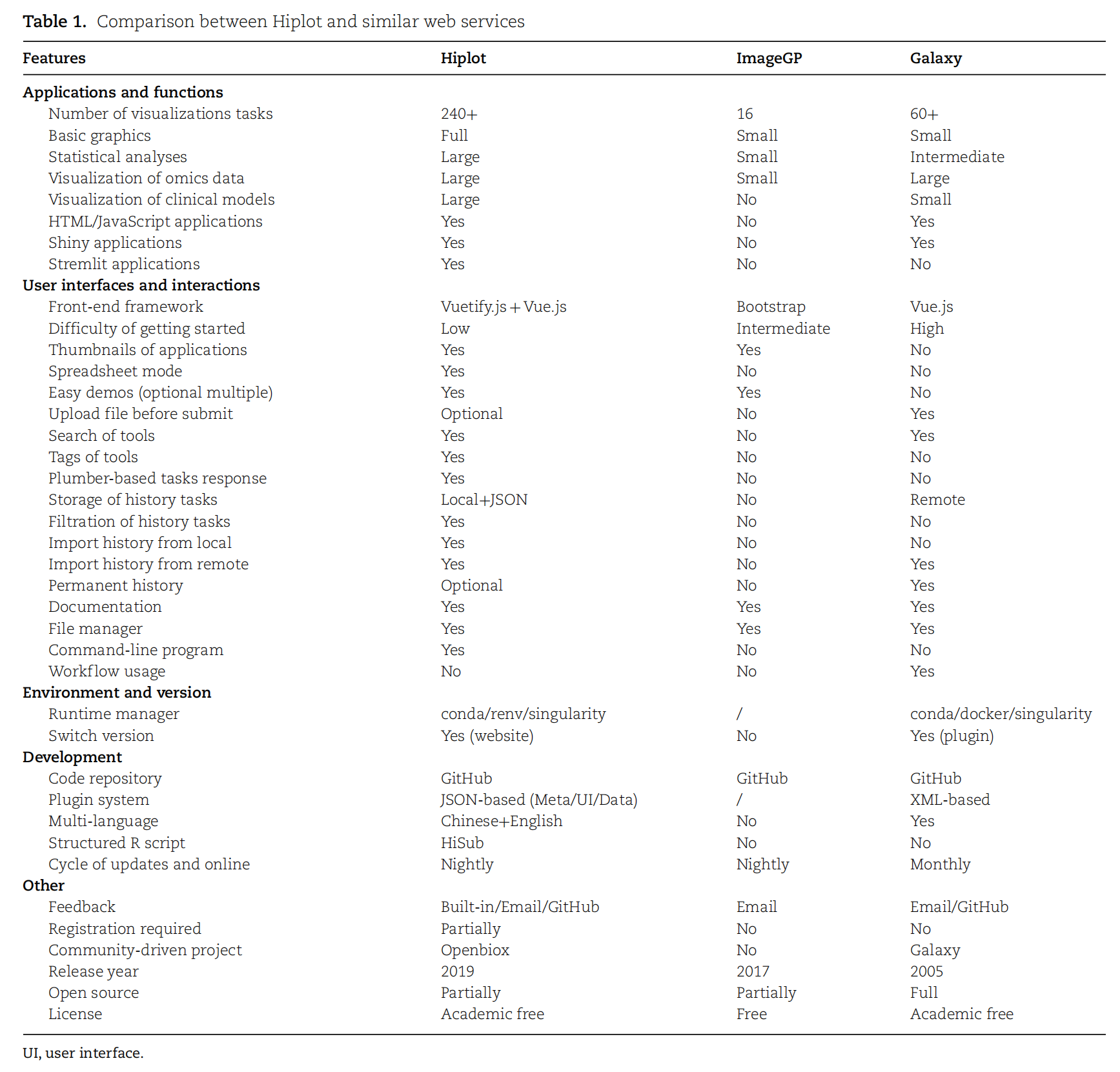
Table 1. Hiplot 與類似 web 服務之間的比較
在 Hiplot 中實現的諸多基礎可視化改進已吸引了科學界的廣泛用戶,同時平臺還提供組學與臨床數據可視化功能,為生物醫學和生物學研究者提供更深入的支持。如今,用戶可在 Hiplot 網站上自由交互式探索癌癥多組學數據集,全面實現基因組結構、染色體分布、遺傳變異、群體遺傳學、基因表達譜、基因通路富集和腫瘤微環境(TME)等多組學數據可視化(Figure 1)。此外,平臺還提供基于機器學習的可視化方法,包括無監督聚類、降維算法(DRAs)、線性/非線性回歸、薈萃分析、生存分析和風險模型等,使用戶能夠關聯多維特征并開展轉化研究(Figure 1)。
除了全面的功能(基礎科學圖表、組學數據可視化、臨床模型)外,Hiplot 的其他主要優勢還包括:簡潔的設計與交互方式(如基于電子表格的數據表)、豐富的輸出樣式與配色主題、基于 Plumber 的多用戶后端任務框架、基于 JSON 的插件系統、便捷的任務復現機制以及功能的實時更新(Figure 1)。Hiplot 采用的現代化用戶界面和高效交互方式顯著降低了用戶學習成本。用戶可通過主網頁客戶端或命令行程序調用 Hiplot 的豐富功能(Figure 2A)。平臺通過標簽搜索的關鍵詞模糊匹配功能,可快速定位基礎工具、高級工具、臨床工具和迷你工具等不同模塊的網頁插件,頂部路徑欄也支持插件切換跳轉。此外,卡片縮略圖能幫助用戶快速識別所需工具。相較于傳統生物信息學網站僅支持文件路徑選擇或文本區域上傳的模式,Hiplot 插件中具有更佳可讀性和可編輯性的電子表格數據編輯器,可能成為同類網站的可選替代方案。同時,海量插件提供的豐富配色、主題和輸出選項,使用戶能輕松生成個性化結果。基于 Plumber 的多用戶響應機制相比需要重新加載依賴項的工作流式響應方法,進一步提升了輕量化可視化任務的執行效率(Figure 2B)。來自本地或遠程存儲的標準 JSON 數據對象可在數秒內復現插件的輸入輸出。本研究實現的基于 JSON 的插件系統,使作者能夠開發這個涵蓋多研究領域的大規模網站——特別是結構化的 UI 組件描述通過提供多插件共享的字段值控制組件/后端功能,簡化了冗余開發工作。網站提供的 JSON 文件還支持用戶在瀏覽器和命令行環境中輕松復現任務。此外,網站功能的實時更新將加速 Hiplot 的開發周期。雖然目前尚未支持工作流編輯器設置任務,但通過命令行程序將 Hiplot 功能整合至現有數據分析流程是可行的。若未來用戶需求強烈,可利用現有插件構建類 Galaxy 界面以實現更優的工作流整合。
?
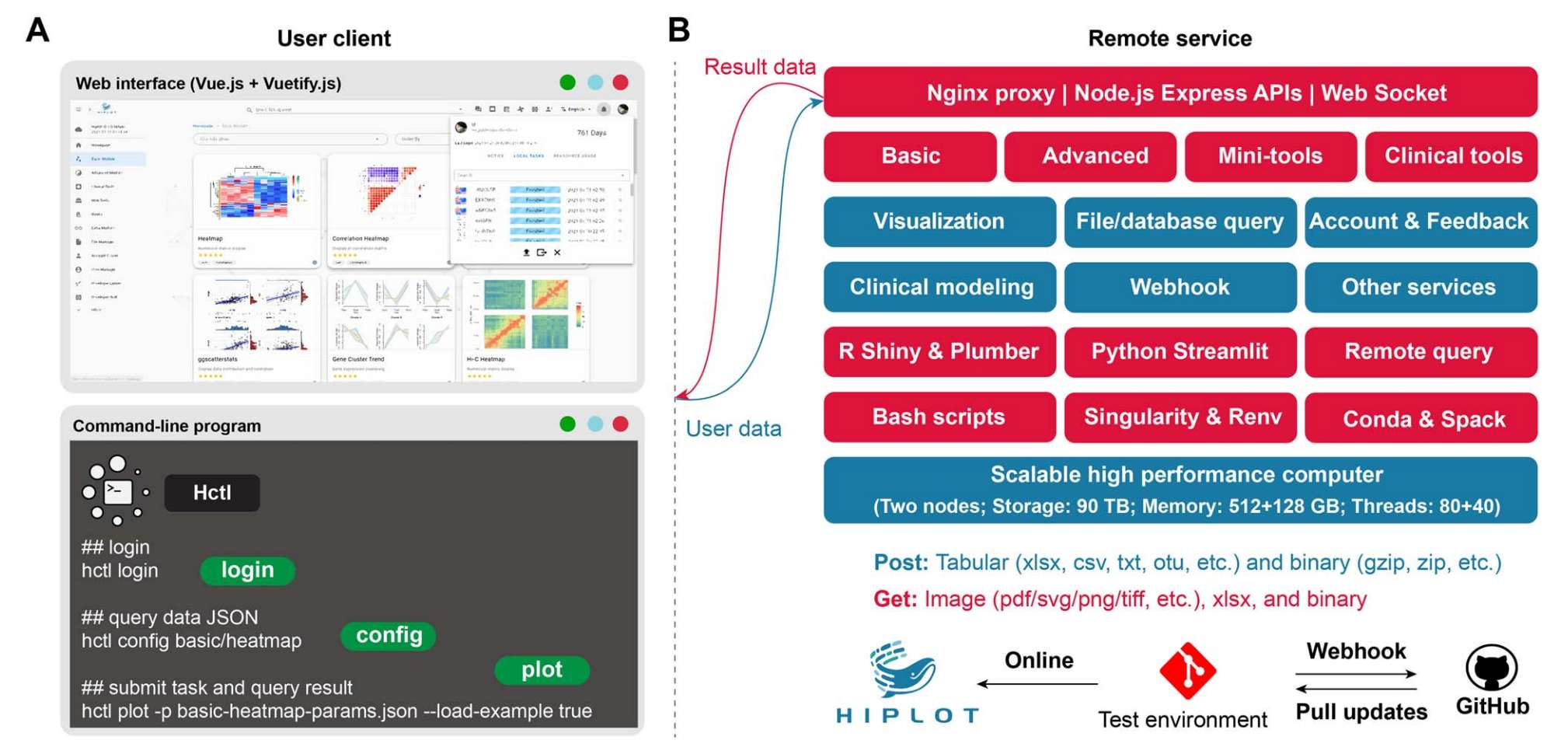
Figure 2. 網站基礎架構和從客戶到后端服務的組件
(A) Hiplot 的網頁客戶端與命令行程序 Hctl。頂部窗口展示的是 Hiplot 網頁客戶端在基礎模塊頁面的截圖,其中列出了若干基礎應用卡片。頁面左側設有主導航菜單,方便用戶在不同模塊間切換瀏覽,同時通知窗口會顯示歷史任務記錄。底部窗口則展示了 Hctl 的子命令功能,包括登錄(login)、配置(config)和提交(submit)。使用 Hctl 程序需先完成登錄,config 子命令用于查詢演示數據/參數,submit 子命令則用于提交任務。
(B) 基礎設施示意圖呈現了 Hiplot 網絡服務的核心后端服務與硬件資源架構。Hiplot 的網頁端和命令行客戶端通過 Nginx proxy / Node.js Express API / Web Socket services 進行通信。平臺的任務插件分布在四大核心模塊:基礎工具、高級工具、迷你工具和臨床工具。除基于 JSON-based Vue.js插件外,還引入了 R Shiny 和 Python Streamlit 框架來構建交互式應用。Hiplot 的運行環境由 conda、spack、Singularity、renv 進行管控,當 Github 上的 Hiplot 或 Openbiox 代碼庫有新提交時,相關插件會自動同步至開發或生產環境進行驗證。
?
2. 使用案例
由于篇幅所限,作者僅基于示例/真實數據集介紹少量具有代表性的功能插件。這將有助于讀者理解網站的功能分類和基本使用方法。這些用例主要分為三大部分:基礎科學圖表、癌癥組學數據可視化以及臨床模型。
使用案例1:基礎科學圖表
作者選取 Hiplot 網站上使用頻率最高的科學圖表——熱圖(Heatmap),來展示基礎可視化任務的常規操作流程(Figure 3A)。用戶進入插件頁面后,可通過點擊頂部/底部的演示按鈕加載示例數據,查看標準輸入格式。需特別注意的是,熱圖插件要求輸入數值型數據表(行代表特征,列代表樣本)及可選的行/列注釋信息。這些數據表支持通過剪貼板粘貼、本地文件上傳或遠程文件服務器三種方式導入。對于超過 2MB 的基因表達矩陣文件,建議使用文件上傳器選擇路徑模式進行傳輸。熱圖插件提供通用參數用于控制圖表寬度、高度、字體、主題、行列注釋的配色方案以及行列文本和標題的字體大小。額外參數還包括熱圖配色、數據標準化方式、高變異特征篩選、聚類方法、距離度量及數值顯示等設置。當前默認采用歐氏距離(Euclidean distance)和 ward.D2 算法進行聚類分析,用戶可嘗試不同聚類方法與距離度量組合以獲得最佳效果。當輸入特征數量龐大時,選擇高變異特征有助于進行子集的無監督層次聚類。若顏色梯度區分度不足,用戶可通過"scale"參數選擇按行或列進行數據標準化。任務提交后,數據流會經過 Base64 編碼傳輸至后端服務(Figure 3B),由可用的 Plumber 工作節點進行處理。最終生成的熱圖結果(如 JPG/PDF 格式)將在底部預覽窗口顯示并提供下載。用戶可將參數導出為本地 JSON 文件以便后續快速加載,登錄用戶還可通過預覽窗口的同步按鈕將結果永久保存至云端文件管理器。
?
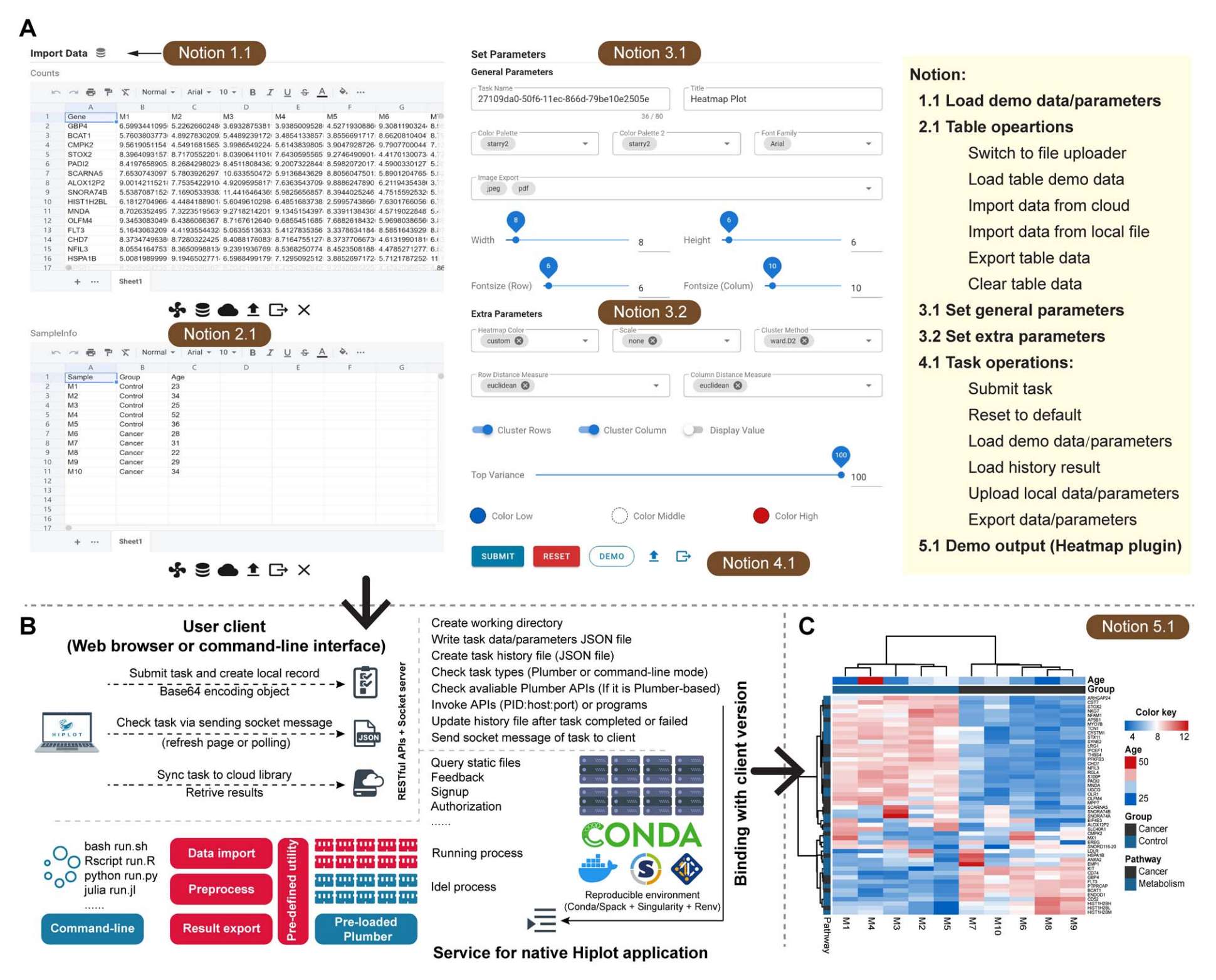
Figure 3. 熱圖插件的使用流程和任務處理步驟
(A) 熱圖插件的網頁界面及提交新任務的主要操作步驟。表格數據可通過電子表格網頁組件進行導入、預覽和編輯。右側面板列出了熱圖的通用參數和高級參數,這些參數可控制數據預處理、聚類步驟及熱圖輸出樣式。其中 "top variance" 參數可用于篩選部分基因進行聚類分析。所有參數均以 JSON 格式存儲,可直接導出用于復現輸入設置。用戶還可通過云端文件管理器的歷史記錄功能重現輸入輸出數據。
(B) 后端處理任務的服務流程示意圖。提交的數據會經過 Base64 編碼,隨后由可用的 Plumber 工作節點執行熱圖插件的核心代碼。任務完成后,后端將返回包含任務歷史信息的 JSON 文件,以及用于獲取圖像和記錄輸出的路徑。
(C) 熱圖插件的示例輸出效果。每列代表一個樣本,每行代表一個基因。示例輸入數據可清晰區分兩個聚類(癌癥組與對照組)。行列注釋分別顯示了基因分類(癌癥相關或代謝相關)和樣本類型(癌癥或對照)。
網站基礎模塊提供了其他基于表格數據的基礎科學圖表,可用于探索相關性、數據比例或拓撲結構、分布特征、演化趨勢、網絡關系和空間特性等,這些圖表均基于 Hiplot 原生框架構建。此外,迷你工具模塊還提供實用的細分功能,可簡化數據表中分類變量與連續變量的統計分析,并支持將 Hiplot 生成的圖表或本地 PDF 文件進行多圖組合。
?
使用案例2:基于組學的癌癥數據可視化
Hiplot 平臺已開發了數十個多組學數據可視化插件,尤其專注于癌癥基因組學和轉錄組學分析,主要包含 Shiny 應用程序和原生 Hiplot 插件。2019 年,Openbiox 社區啟動了開源項目 UCSCXenaShiny(Figure 4A),該項目通過 R 函數和基于 Shiny 的網頁界面,使用戶能夠搜索、下載、探索并快速分析/可視化來自 UCSC Xena 數據中心的基因組數據集。作者采用 UCSCXenaShiny 的泛癌模塊分析表明,促甲狀腺激素釋放激素的高表達與癌癥基因組圖譜(TCGA)數據庫中急性髓系白血病患者(預后改善)和膠質瘤患者(預后較差)均存在顯著關聯(Figure S10)。該交互式應用還能探索其他任何基因表達異常或存在序列突變的基因在主要癌癥類型中的預后意義。用戶可通過點擊 Hiplot 網站高級模塊中的"Shiny"及相關標簽,發現更多基于 Shiny 的癌癥組學可視化應用,例如全基因組關聯研究相關的 Shiny 插件。
?
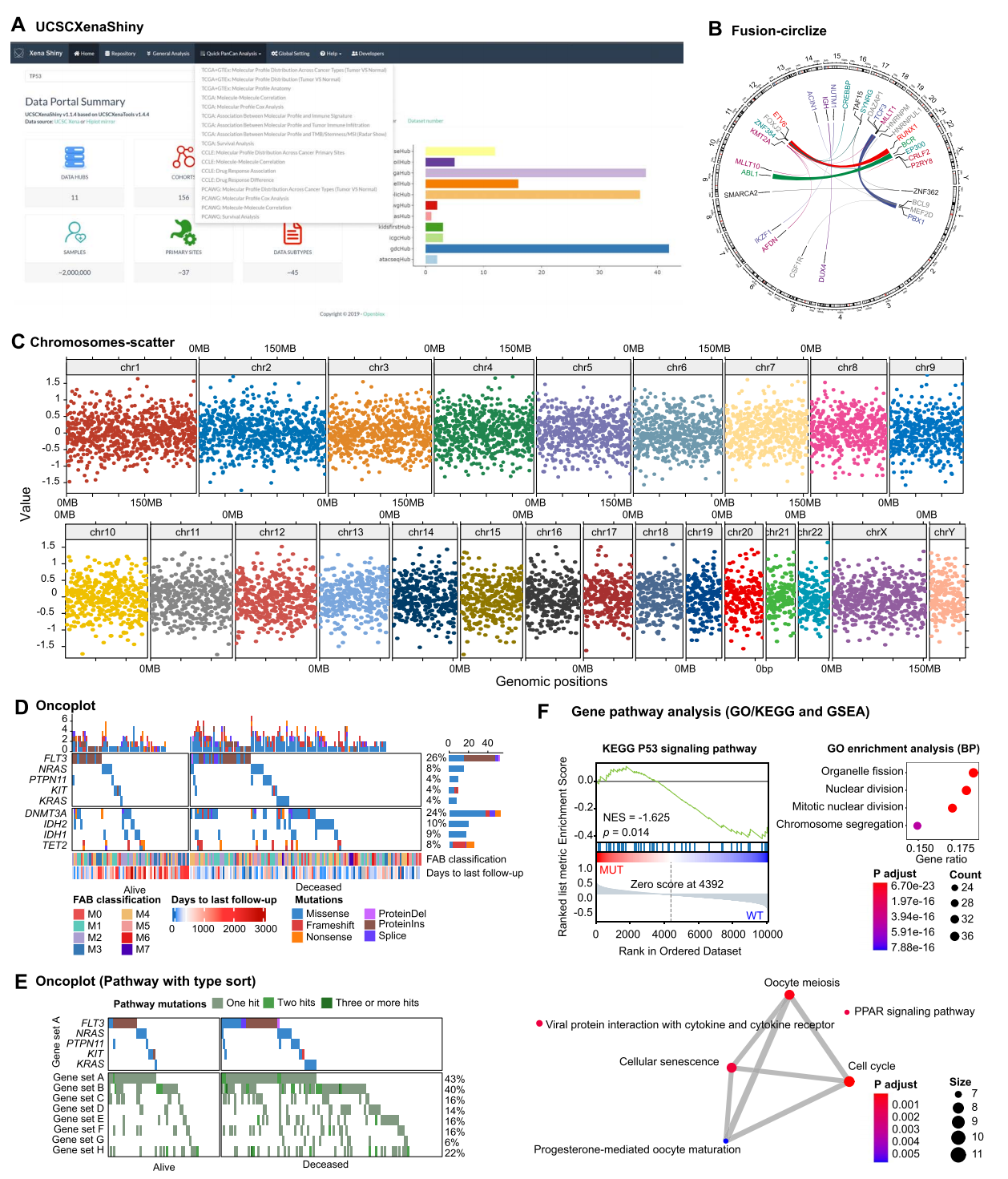
Figure 4. Hiplot 中基于組學的可視化功能的代表性用例
(A) 圖為 UCSCXenaShiny 應用程序界面截圖,該基于 R Shiny 開發的應用可交互式挖掘 UCSC Xena Hub 的公開數據集。
(B) fusion-circlize 插件的示例輸出。展示了 B 細胞前體急性淋巴細胞白血病 (BCP-ALL) 中已知的基因融合事件,包括 ETV6::RUNX1、BCR::ABL1、DUX4 fusions、ZNF384 fusions、MEF2D fusions 和 KMT2A fusions 等。染色體按順時針方向排列,基因融合事件通過彩帶連接不同染色體,連接線及文本的顏色與寬度分別代表融合基因類別和出現頻率。
(C) chromosomes-scatter 插件的示例輸出。不同顏色表示各染色體上的模擬數值分布。
(D) Oncoplot 展示 TCGA 數據庫中急性髓系白血病隊列的選定突變基因,患者按生存狀態分為兩組,不同突變類型以顏色區分,底部方框顯示患者元注釋信息。
(E) 另一個 Oncoplot 通過添加基因通路行和患者數據實現突變分類可視化。
(F) gsea 和 clusterprofile-go-kegg 插件的示例輸出:GSEA 結果以 PDF 格式呈現,顯示標準化富集分數(NES)和 P 值;clusterprofile-go-kegg 插件則通過氣泡圖和網絡圖展示基于示例數據的 GO 通路富集分析結果。
除基于 Shiny 的應用程序外,Hiplot 平臺還提供多個基于 JSON 的原生插件用于交互式可視化大規模癌癥組學數據。例如:融合基因環狀圖插件 (fusion-circlize) 在染色體層面展示 B 細胞前體急性淋巴細胞白血病 (BCP-ALL) 中的典型基因融合事件,包括 RhoGEF 與 GTPase (BCR)-ABL proto-oncogene 1、ETS 變異轉錄因子 6 (ETV6)-RUNX 家族轉錄因子 1 (RUNX1)、雙同源框基因 4 (DUX4) 融合、鋅指蛋白 384 (ZNF384)融合、肌細胞增強因子2D(MEF2D)融合、組氨酸甲基轉移酶2A(KMT2A)融合以及睪丸核蛋白(NUTM1)融合等 (Figure 4B)。染色體散點圖插件 (chromosomes-scatter) 通過染色體坐標散點展示數值分布 (Figure 4C),可用于全基因組尺度下基因拷貝數和表達水平的比較分析。而腫瘤突變圖譜插件 (oncoplot) 則可視化 TCGA 急性髓系白血病(LAML)隊列中患者特異性突變基因與通路信息 (Figure 4D and E)。discover-mut-test 插件通過互斥共現分析算法,在識別基因互斥事件方面表現出優于傳統 Fisher 精確檢驗的性能。基于已發表的 BCP-ALL 患者變異數據,作者使用多個 Hiplot 插件證實 BCP-ALL 中主要融合基因與染色體異常多呈互斥關系,但細胞因子受體樣因子2(CRLF2)融合、DUX4融合和BCR-ABL1突變會與Janus激酶2(JAK2)、堿性螺旋-環-螺旋(bHLH)轉錄因子原癌基因(MYC)及RUNX1序列變異共存 (Figure S11)。
轉錄組數據可通過基礎與高級插件(如熱圖、一致性聚類、復合熱圖、箱線圖、火山圖和偽增強MA圖等)進行無監督聚類和表達水平可視化。一致性聚類插件 (cola) 基于基因表達數據完成共識聚類演示 (Figure S12)。該插件通過數據采樣整合多種高變異基因篩選與聚類方法,提供獲取穩定分群的接口。復合熱圖插件 (complex-heatmap) 則可關聯基因表達、基因突變與臨床特征等多組學數據 (Figure S13)。獲得亞群差異表達基因 (DEGs) 后,用戶可在火山圖中交互式添加目標基因標簽。
clusterprofiler-go-kegg 插件支持基于多列數據一次性完成多組通路富集分析 (Figure 4F),可接收基因符號、Ensembl ID 等多種標識符輸入,并將多圖整合輸出為 PDF 和 Excel 文件。基因集富集分析 (GSEA) 插件完整復現了 Broad 研究所命令行版本的功能,相比桌面版更支持同時比較多個亞群與基因集 (Figure 4G)。此外,作者重構了 immunedeconv R 包的網頁界面,支持基于多種算法進行腫瘤微環境分析和免疫細胞組分計算 (Figure S14)。最后,作者采用主成分分析、t-SNE、UMAP 和自組織映射等降維方法可視化經典鳶尾花數據集,展示數值特征在降維空間中的已知分類分布 (Figure S15)。這些方法可根據特征排序或距離度量從多維組學數據中篩選重要維度作為代表性數據,有助于識別具有生物學/臨床意義的潛在新細胞/患者亞型。
?
使用案例3:臨床模型
臨床模型可視化功能可用于識別疾病風險因素并構建回歸模型的典型圖表。本節提供五個應用案例。首先,作者使用 ezcox 插件在臨床數據可視化任務中執行 Cox 回歸分析,構建包含控制變量的多變量生存風險模型(Figure 5A and B);metawho 插件是"治療獲益人群 Meta 分析方法"的簡易網頁實現,用于開展 Meta 分析任務(Figure 5C);risk-plot 插件(Figure 5D)和 survival 插件(Figure 5E)分別用于可視化患者疾病風險模型與生存數據。risk-plot 插件展示生存狀態與風險因素相關性,并按風險評分從低到高排序患者;最后,作者基于 survival 包中的肺癌數據集,通過 nomogram 插件(Figure 5F)建立預測性風險評分的演示模型。
?
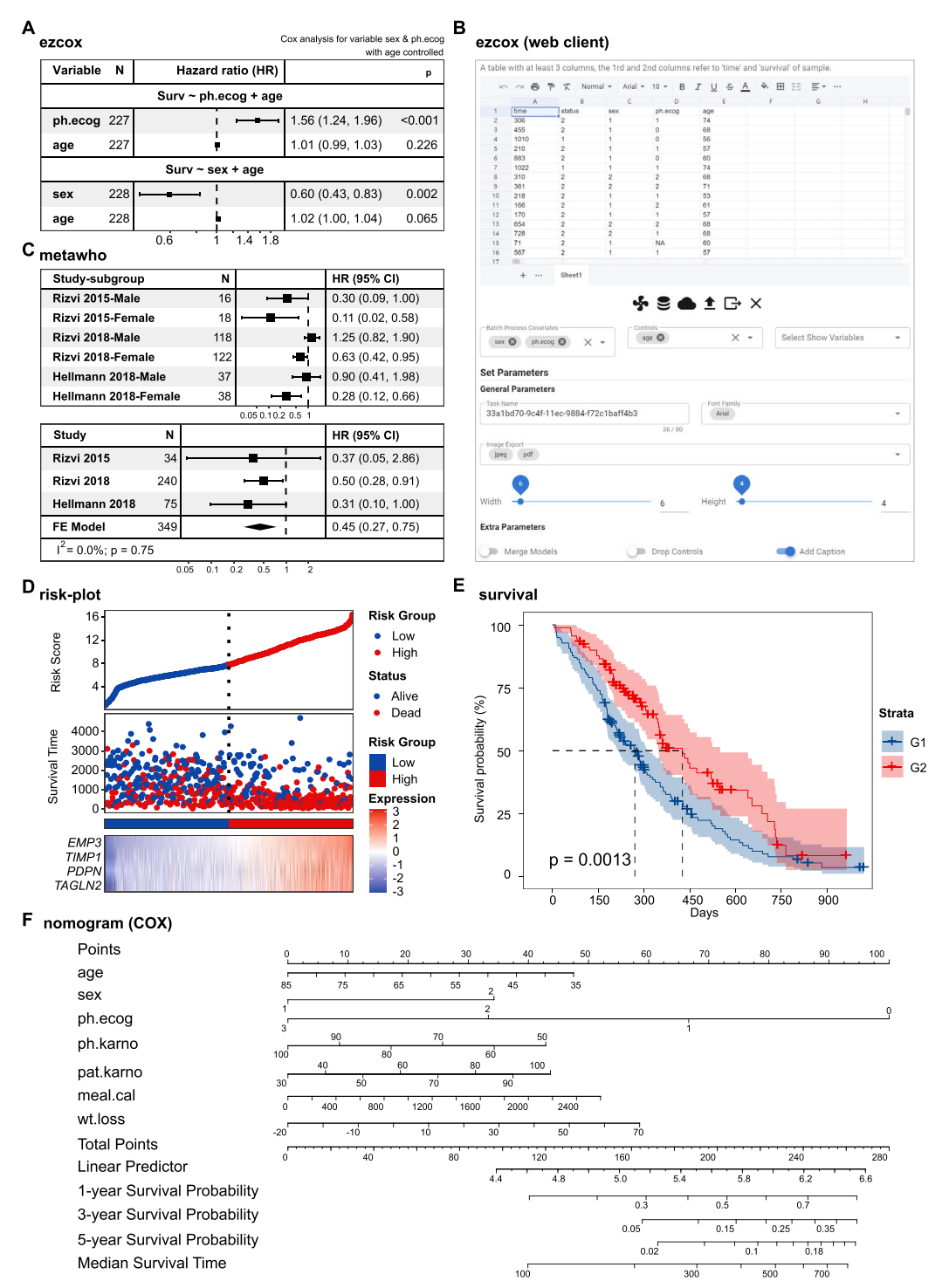
Figure 5. Hiplot 中臨床數據可視化的代表性用例
(A-B) 分別展示了使用 survival 包中肺癌數據集的森林圖示例及 ezcox 插件的網頁界面。Cox 模型顯示,當以年齡作為控制變量時,性別(sex)和 ECOG 評分(ph.ecog)分別與高風險(HR: 1.56, CI: 1.24–1.96)和低風險(HR: 0.60, CI: 0.43–0.83)顯著相關。
(C) metawho 插件的示例輸出,表格中展示了各亞組研究和整體研究的風險比(HR)。
(D) risk-plot 插件的示例結果。根據風險評分中位數將患者分為高風險(紅色)和低風險(藍色)亞組,每個點代表一名患者。中間區域紅色點表示死亡患者,底部區域則顯示特征基因表達水平,包括上皮膜蛋白3(EMP3)、金屬蛋白酶組織抑制劑1(TIMP1)、足萼蛋白(PDPN)和轉膠蛋白2(TAGLN2)。
(E) survival 插件的模擬數據輸出,展示 3 年生存曲線。
(F) 基于 survival 包肺癌數據集構建的列線圖(nomogram)預測模型,P 值通過對數秩檢驗計算得出。HR:風險比;CI:置信區間。
?
3. 用戶任務和基準測試顯示基于本機 Hiplot 插件的性能
作者收集了 2022 年 4 月 18 日至 24 日期間基礎工具、高級工具和迷你工具插件的運行耗時數據,并進行了可視化基準測試,為后續可能的優化提供參考。數據顯示,基礎模塊和高級模塊中分別有超過 80% 的插件可在 5 秒內和 3 分鐘內完成分析(Figure 6A)。此外,用戶提交的任務表明,大多數用戶傾向于上傳小于 10MB 的文件至本網站進行可視化分析(Figure 6B)。在耗時超過 1 分鐘的插件中(至少 10 次任務記錄),世界地圖插件的中位耗時最長,而熱圖和相關性圖則是使用頻率最高的兩個插件(Figure 6C)。
?
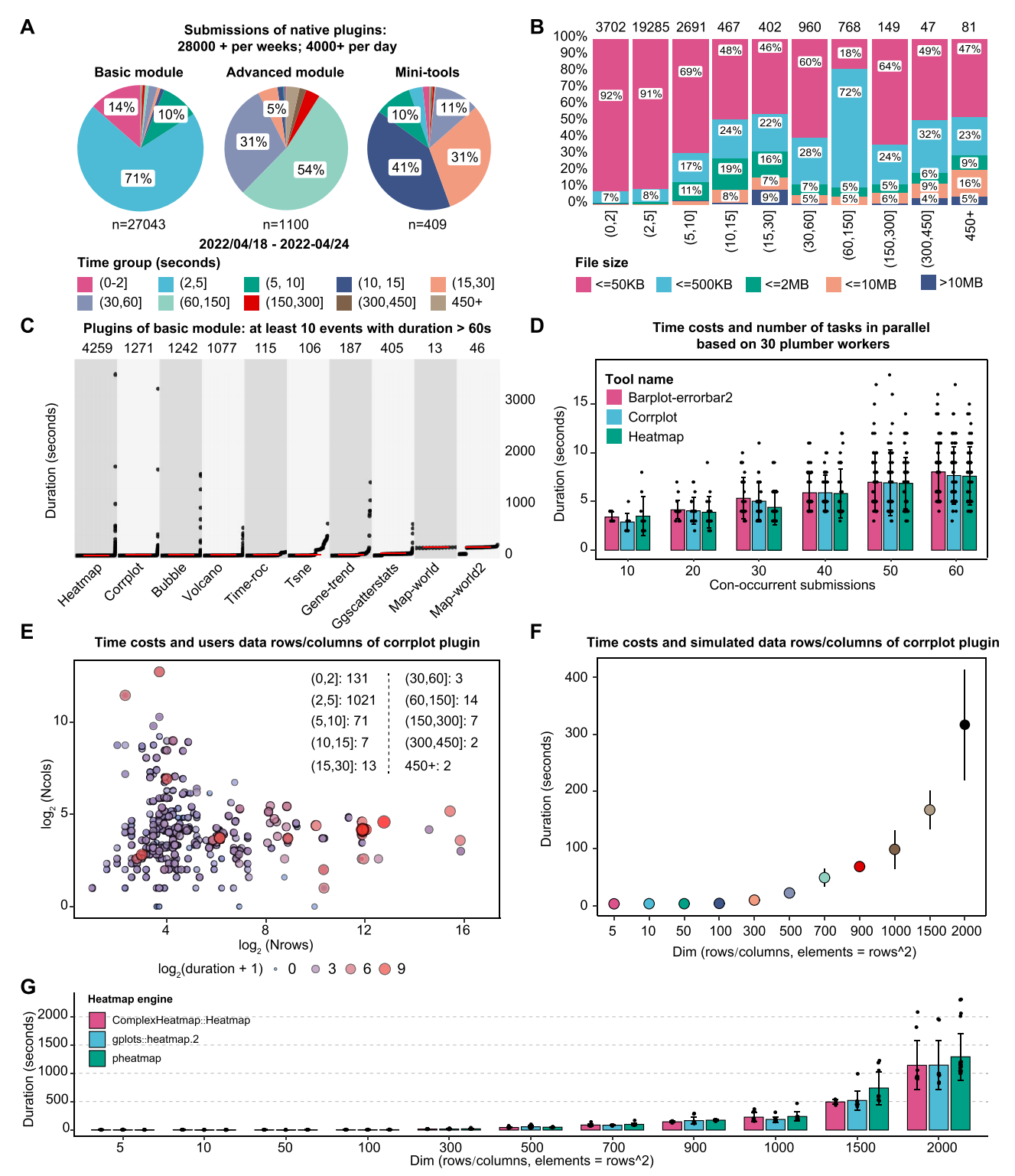
Figure 6. 用戶任務和選定本機插件的基準測試顯示了 Hiplot 的性能
(A) 餅圖展示 2022 年 4 月 18 日至 24 日一周內基礎工具、高級工具和迷你工具插件的耗時分布,不同顏色代表不同耗時區間組別。
(B) 條形圖顯示任務數據量與耗時的關系,不同顏色表示不同文件大小分組。
(C) 散點圖呈現基礎模塊中耗時超過 60 秒且執行次數 ≥10 次的任務,插件按中位耗時從低(左)到高(右)排序,面板頂部標注總提交次數。
(D) 帶誤差點的條形圖說明并行提交對任務耗時的影響,barplot-errorbar2、corrplot 和 heatmap 插件用不同顏色區分。
(E) 氣泡圖展示任務耗時與數據行列數的正相關關系。
(F) 誤差棒散點圖顯示 corrplot 插件在不同數據維度下的耗時表現。
(G) 條形圖比較不同熱圖引擎處理不同維度數據時的耗時差異。
作者進一步通過調用并行任務進行基準測試。結果顯示,熱圖(heatmap)、相關性圖(corrplot)和誤差棒條形圖(barplot-errorbar2)等插件的并行任務數量與耗時呈正相關。不過,在 10 至 60 個并行提交的不同測試條件下,大多數任務仍能在 15 秒內完成(當前 Hiplot 實際并發數小于 10)(Figure 6D)。數據行列數是影響耗時的另一關鍵因素。以相關性圖插件為例,雖然大部分提交可在 5 秒內完成,但行列數增加會顯著延長處理時間(Figure 6E)。基于不同維度數據矩陣的標準測試表明,當數據超過 1000 行/列(即 100 萬個元素)時,相關性圖插件的性能開始下降(Figure 6F)。采用不同 R 語言熱圖引擎實現的相關性熱圖插件也呈現相似規律(Figure 6G)。
?
4. 用戶訪問代表受歡迎程度和潛在影響
Hiplot 項目于 2019 年啟動,首個正式版本于 2021 年 3 月發布。作者對 2020 年 7 月 9 日至 2021 年 12 月 31 日的網站訪問數據統計顯示:該平臺網頁端已獲得來自全球 100 個國家超 250 萬次訪問,日均訪問量超 5000 次,日均任務提交量達 3000 次(Figure S16A)。平臺注冊用戶數突破 2.2萬(Figure S16B),其中 160 余個插件訪問量逾千次(Figure S16C)。基礎熱圖插件單模塊訪問量超 7 萬次,相關性熱圖、氣泡圖、箱線圖、線性回歸和火山圖等核心插件訪問量均突破 2 萬次。基于 clusterProfiler 和 GSEA 的通路分析工具分別實現 2.1 萬次和 5000 次調用,高級模塊中的 UCSCXenaShiny 應用訪問量達 6300 次,迷你工具模塊的 PDF 拼圖插件使用超 7000 次。這些流量增長數據印證了平臺部署的各類網絡插件所產生的學術影響力。
?
討論
當今生物學/生物醫學與計算科學的聯系比數十年前更為緊密。多維數據可視化技術(如現代統計圖表和組學數據可視化)已成為生物醫學數據挖掘不可或缺的工具。目前研究人員主要通過三種途徑實現生物醫學數據可視化:傳統商業桌面軟件、編程語言/庫以及網絡工具。然而,繪制高質量、可直接出版的科學圖表仍是生物信息學家和專業數據分析師面臨的挑戰,他們通常需借助 R、Python 等編程語言庫來完成,這對缺乏編程技能的生物學家和臨床醫生更為困難。因此,基于現代網絡技術的生物醫學可視化工具正日益受到科研界青睞。
現有基于云計算的平臺主要聚焦上游組學分析流程,對輕量級科學數據可視化和交互式分析的支持不足,且缺乏實時更新能力。這些平臺往往操作復雜,在處理大數據時步驟冗余,其網頁插件開發模式難以友好支持基于表格的輕量級可視化任務。科研用戶對現代科學圖表系統實現和交互式數據挖掘應用的高頻需求,對推動生物醫學領域數據科學發展具有重要意義。
本研究提出的 Hiplot 云服務平臺,為生物醫學數據交互式可視化提供了全面易用的解決方案。其簡潔的用戶界面(如基于電子表格的數據導入)和高效交互設計,極大降低了非編程用戶的輕量級可視化任務學習成本。作者通過支持版本控制的多用戶 Plumber 服務器和基于 JSON 的結構化插件系統,加速了輕量級科學可視化插件的開發。自 2019 年以來,Hiplot 聯盟已實現數百個生物醫學可視化插件。本研究不僅提供了該綜合平臺的用戶使用全景數據,還通過精選原生插件進行基準測試,這些工作將促進 Hiplot 及相關工具的迭代優化。盡管該網站在處理大數據和上游組學分析方面仍有改進空間,但現有可視化插件已為生物醫學數據挖掘提供了重要在線資源。未來平臺將擴展更多可視化功能和公共生物醫學數據集的交互式挖掘。這一新興工具包將持續惠及缺乏編程技能的生物信息學、生物醫學及其他領域的數據科學家。
?
--------------- 結束 ---------------
?
注:本文為個人學習筆記,僅供大家參考學習,不得用于任何商業目的。如有侵權,請聯系作者刪除。
?

?
)












)





