在轉錄組 Bulk 測序數據分析中,熱圖是展示基因表達模式、樣本聚類關系的核心可視化工具。一張高質量的熱圖不僅能清晰呈現數據特征,更能提升研究成果的展示效果。本文基于 R 語言的pheatmap包,整理了六種適用于不同場景的熱圖繪制方法,涵蓋基礎聚類、分組對比、通路注釋等需求,私信即可獲取全部代碼,方便科研人員快速實現數據可視化。
一、繪圖前的數據準備
熱圖繪制的核心是基因表達矩陣,數據格式的規范性直接影響后續分析效果,需提前做好以下準備:
1.1 表達矩陣格式要求
表達矩陣需滿足:
- 第一列為基因名(后續將作為行名);
- 從第二列開始為樣本的基因表達值(FPKM、TPM 或 Counts 均可);
- 第一行為樣本名稱(作為列名)。
示例格式如下(簡化版):
| GeneSymbol | Sample1 | Sample2 | ... | Sample60 |
|---|---|---|---|---|
| GeneA | 12.3 | 15.6 | ... | 8.9 |
| GeneB | 3.2 | 2.1 | ... | 5.7 |
1.2 數據讀取與預處理
使用read.csv函數讀取數據,注意保留基因名作為行名,避免樣本名被自動修改:
# 讀取表達矩陣(替換為實際文件路徑)
data <- read.csv("gene_expression_matrix.csv", row.names = 1, check.names = FALSE)
# check.names = FALSE:避免樣本名中的特殊字符(如“-”“.”)被自動轉換
1.3 樣本分組注釋
根據研究設計定義樣本分組(如正常組 / 疾病組、處理組 / 對照組等),用于后續熱圖的分組標記:
# 示例:30個正常組樣本 + 30個疾病組樣本(需根據實際樣本數量修改)
sample_groups <- data.frame(Condition = factor(c(rep("Normal", 30), rep("Disease", 30))), # 分組名稱row.names = colnames(data) # 樣本名與表達矩陣列名對應
)
二、六種熱圖繪制方法
2.1 經典聚類熱圖(部分代碼)
適用場景:初步展示基因表達模式與樣本聚類關系,適合探索性分析。
通過行(基因)標準化消除表達量量級差異,結合聚類樹展示基因與樣本的相似性:
library(pheatmap)
library(RColorBrewer)pheatmap(data,color = colorRampPalette(rev(brewer.pal(11, "RdBu")))(100), # 紅藍漸變配色(表達量高→紅,低→藍)scale = "row", # 按行(基因)標準化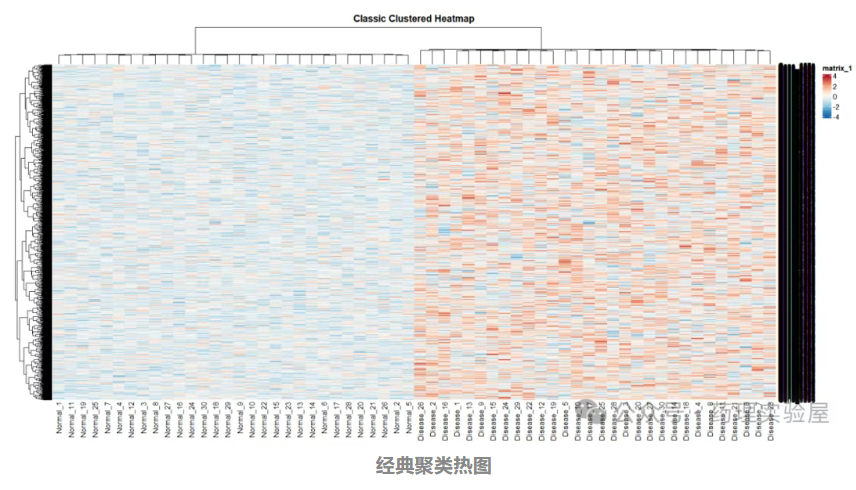
特點:通過顏色梯度直觀展示基因表達高低,聚類樹反映樣本 / 基因的相似性。
2.2 分組注釋熱圖(部分代碼)
適用場景:需突出樣本分組信息(如正常 / 疾病、處理 / 對照),增強組間差異的可視化效果。
在經典熱圖基礎上添加樣本分組注釋條,通過顏色區分不同組別:
# 定義分組注釋顏色(可根據需求調整色值)
annotation_colors <- list(Condition = c(Normal = "#4DBBD5", # 正常組:淺藍色Disease = "#E64B35") # 疾病組:紅色
)pheatmap(data,color = colorRampPalette(rev(brewer.pal(11, "RdBu")))(100),scale = "row",
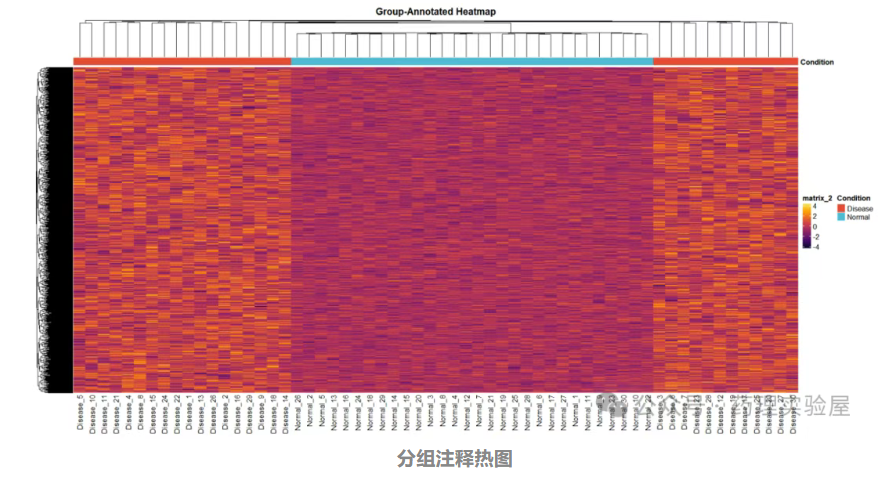
特點:通過頂部注釋條清晰區分樣本組別,便于觀察組內聚類一致性與組間差異。
2.3 二元對比熱圖(部分代碼)
適用場景:需突出基因在兩組間的表達差異(如上調 / 下調),適合差異基因集的可視化。
使用多色漸變(藍→白→紅)強化表達量極端值的對比:
pheatmap(data,color = colorRampPalette(c("#2c7bb6", "#abd9e9", "#ffffbf", "#fdae61", "#d7191c"))(100),# 顏色梯度:深藍(低表達)→淺藍→白→淺紅→深紅(高表達)scale = "row",annotation_col = sample_groups,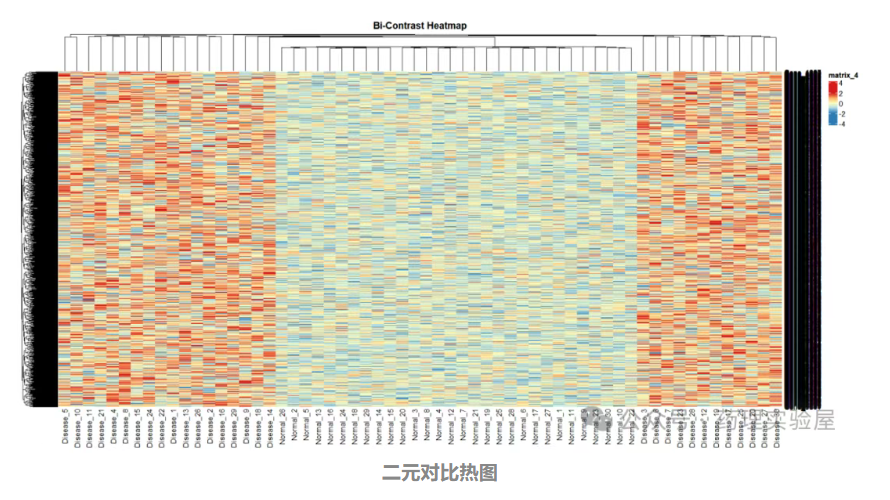
特點:中間白色為基線,兩端顏色強化高低表達差異,適合直觀展示基因在兩組中的表達趨勢。
2.4 子集熱圖(前 20 個基因)(部分代碼)
適用場景:聚焦核心基因(如 top 差異基因、關鍵通路基因),避免全基因集熱圖的冗余。
對基因集進行篩選(如取前 20 個),簡化熱圖復雜度:
library(viridis) # 需提前安裝:install.packages("viridis")# 取前20個基因(可替換為按表達量/差異倍數篩選的基因)
top20_data <- data[1:20, ]pheatmap(top20_data,color = viridis(100), # 青綠色系漸變(美觀且適合色盲友好)scale = "row",
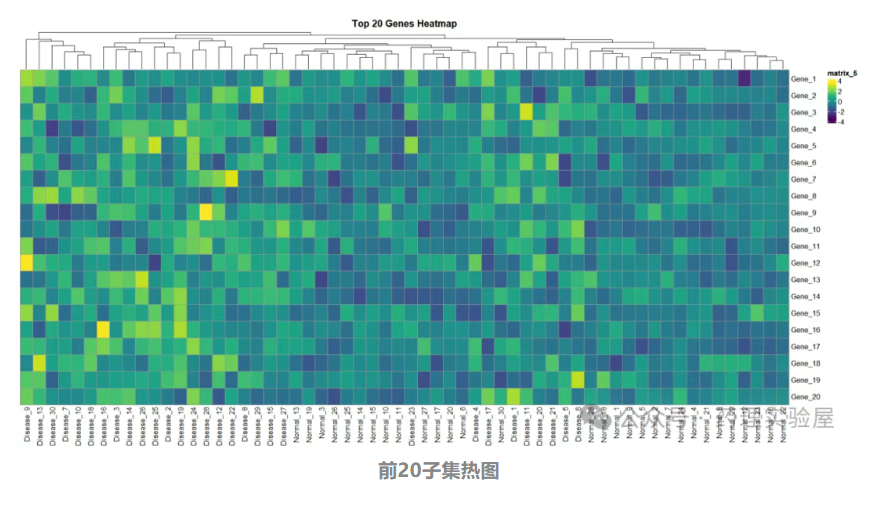
特點:聚焦關鍵基因,減少視覺干擾,適合突出核心基因的表達模式。
2.5 相關系數熱圖(部分代碼)
適用場景:分析樣本間或基因間的表達相關性,探索數據內部關聯性。
基于表達矩陣計算相關系數(如 Pearson),再繪制熱圖:
# 計算樣本間相關系數矩陣(列之間的相關性)
cor_matrix <- cor(t(data)) # t()轉置:樣本為行,計算樣本間相關pheatmap(cor_matrix,color = colorRampPalette(rev(brewer.pal(9, "YlOrRd")))(100), # 黃→紅:相關性從低到高
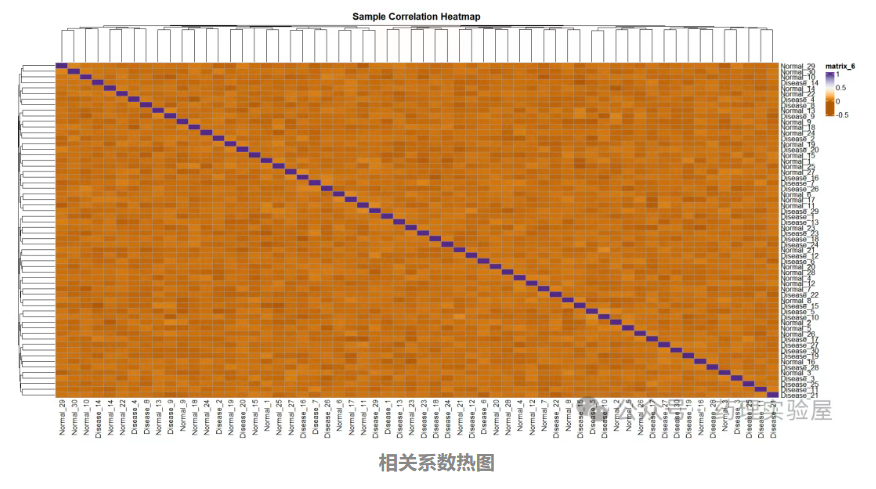
特點:顏色越深表示相關性越高,可用于驗證樣本重復性(組內樣本相關性應高于組間)。
2.6 通路注釋熱圖(部分代碼)
適用場景:展示基因所屬功能通路(如 KEGG、GO 通路),關聯表達模式與生物學功能。
需提前定義基因 - 通路對應關系,添加行注釋(通路信息):
# 定義基因通路
gene_groups <- data.frame(Pathway = factor(sample(c("Pathway A", "Pathway B", "Pathway C"), nrow(data), replace = TRUE)),row.names = rownames(data) # 基因名與表達矩陣行名對應
)# 定義通路注釋顏色
pathway_colors <- list(Pathway = c("Pathway A" = "#7FC97F", "Pathway B" = "#BEAED4", "Pathway C" = "#FDC086")
)
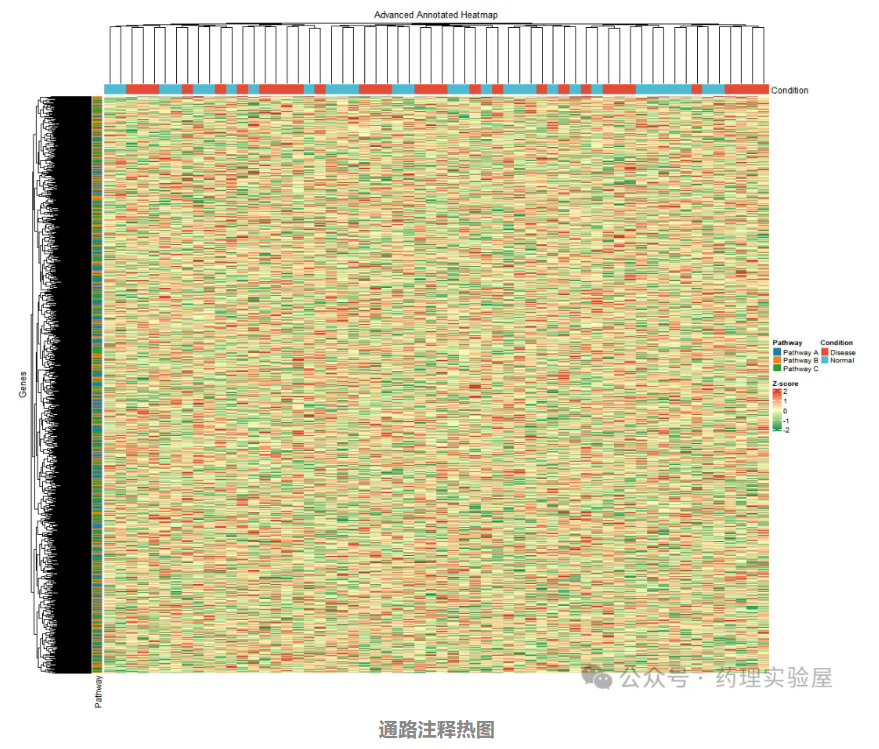
特點:左側添加通路注釋條,可觀察同一通路基因的表達模式一致性。
三、注意事項與優化建議
數據預處理:熱圖對異常值敏感,建議先通過
scale參數(行 / 列標準化)消除量綱影響,或對表達矩陣進行 log2 轉換(若數據未標準化)。參數調整:
show_rownames/show_colnames:基因 / 樣本數量過多時建議設為FALSE,避免標簽重疊;treeheight_row/treeheight_col:調整聚類樹高度,減少冗余空間;fontsize:調整標簽字體大小,優化可讀性。
圖片保存:通過
filename參數保存高清圖片:pheatmap(..., filename = "heatmap.png", width = 10, height = 8, dpi = 300)
以上六種熱圖覆蓋了轉錄組數據分析中常見的可視化需求,可根據實際數據特點(如基因數量、分組類型)選擇合適的方法。代碼中涉及的參數可根據具體需求進一步調整,若有疑問或優化建議,歡迎留言交流。

)


![[java 常用類API] 新手小白的編程字典](http://pic.xiahunao.cn/[java 常用類API] 新手小白的編程字典)














)