
1. Boltz-2介紹
?文章來源:http://jeremywohlwend.com/assets/boltz2.pdf
開源代碼來源:https://github.com/jwohlwend/boltz
該AI模型由麻省理工學院計算機科學與人工智能實驗室與上市AI制藥公司Recursion一起開發,雙方在Boltz-1的基礎之上,通過改進和拓展性能而來。
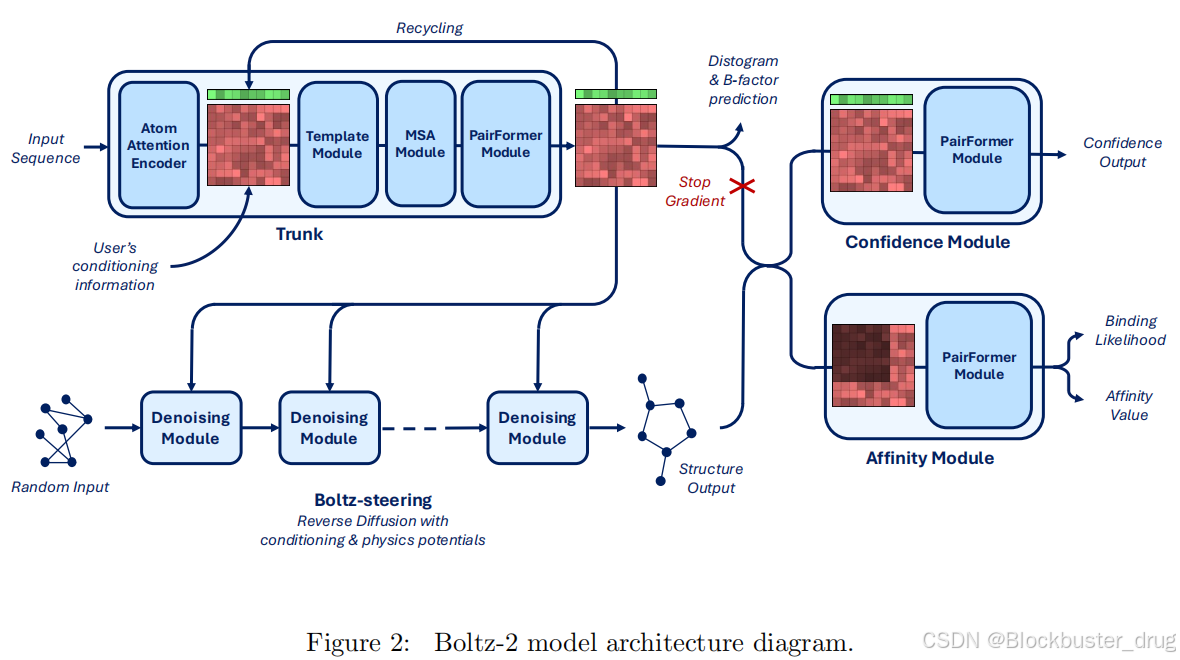
簡單來說,Boltz 與 AlphaFold3 一樣,均是一種全原子共折疊模型,它將蛋白質折疊或結構預測的概念擴展到DNA、RNA、配體中。該模型不僅可以預測分子相互作用的 3D 結構,還可用于分子設計等下游任務。Boltz-2將親和力預測與結構建模相結合,提高了預測結構的物理真實感。
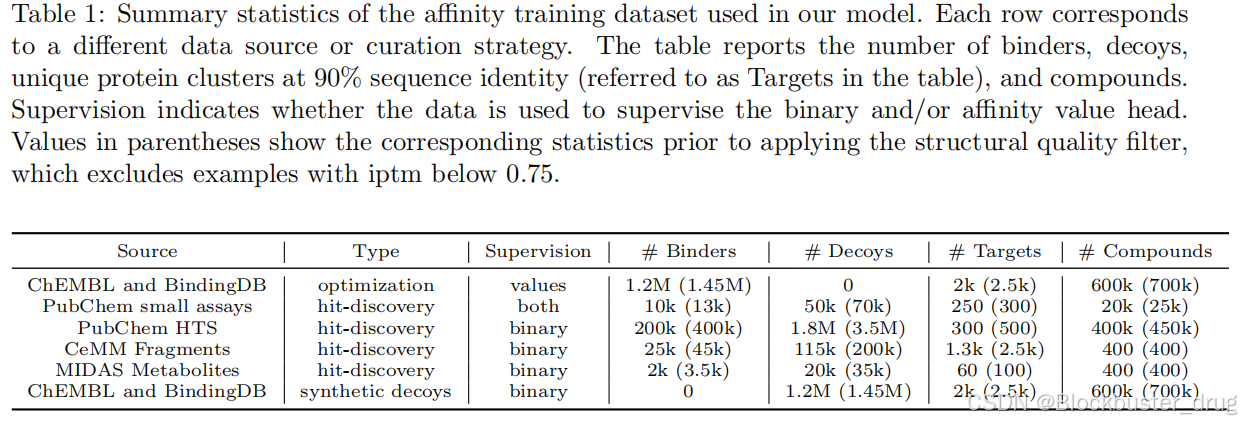
Boltz-2 在一個大型數據集上進行了訓練,該數據集結合了500萬個結合親和力測量值、分子動力學模擬和蒸餾數據,這些方法顯著提高了預測結構的物理真實感。
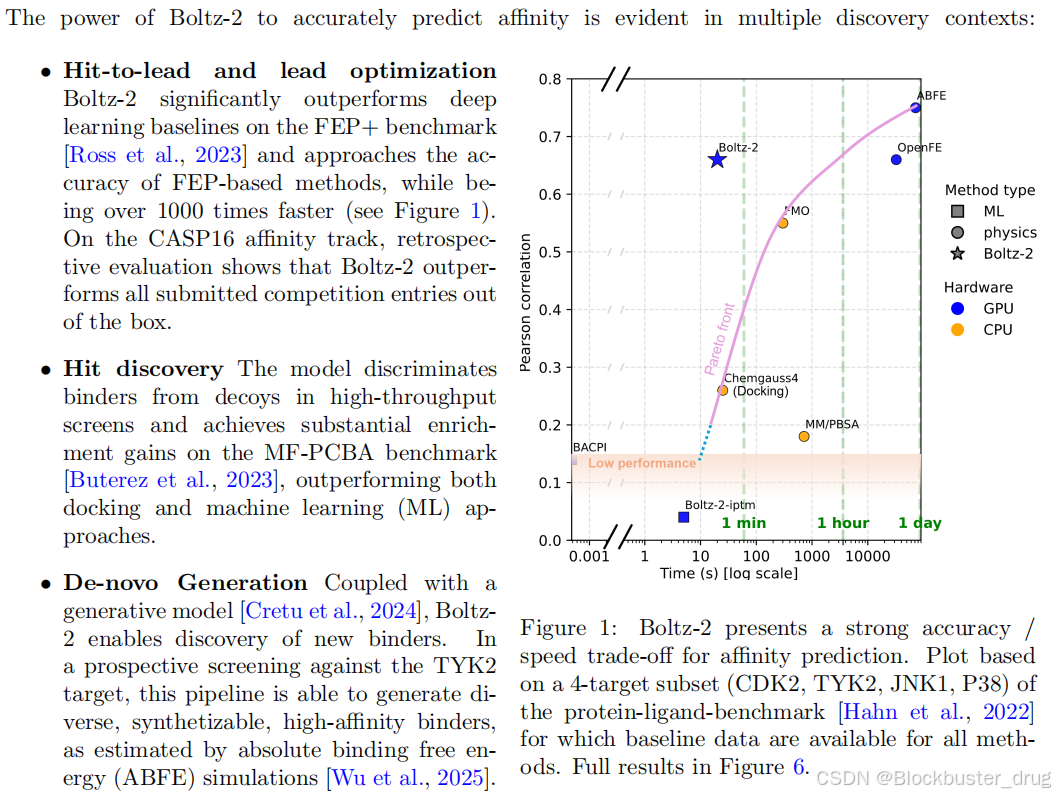
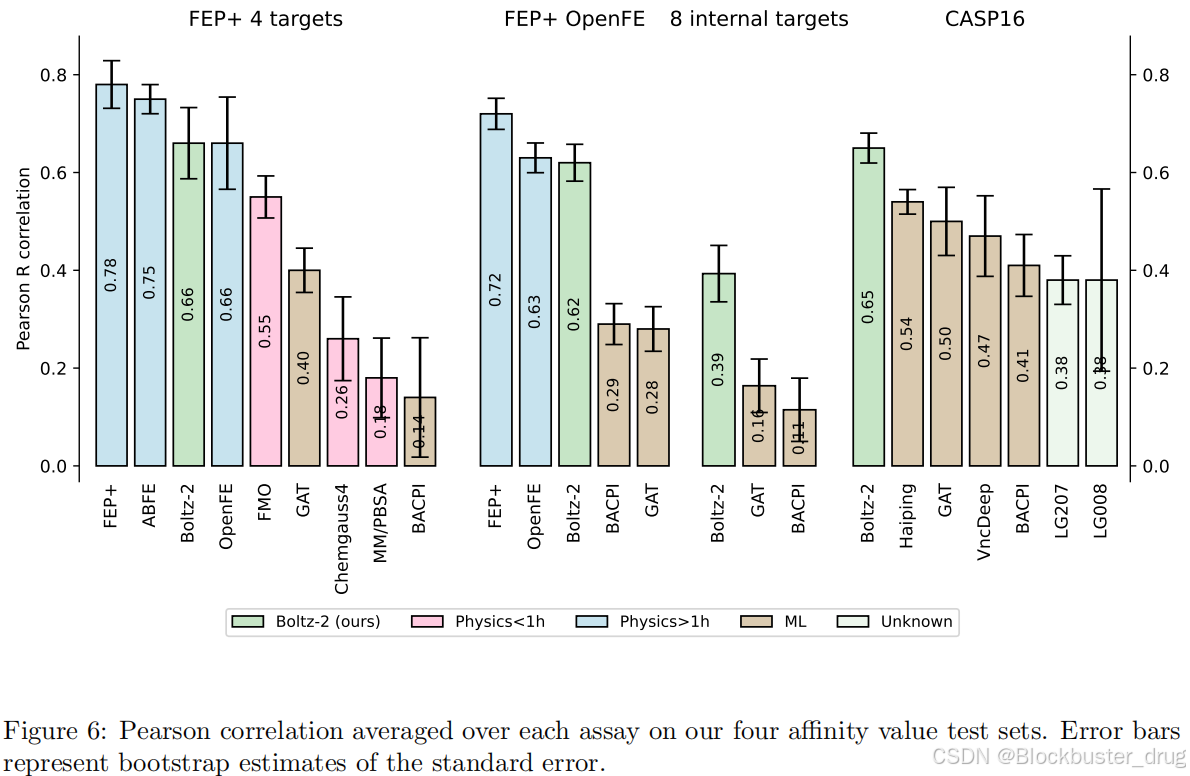
在標準 FEP+ 親和力基準測試中,Boltz-2 實現了 0.62 的平均 Pearson相關系數,能夠與可開源 FEP 流程 OpenFE 相媲美。但在速度方面,Boltz-2 只需 20 秒即可計算出結合親和值,比當前FEP預測快1000倍!
在表示學習方面,親和力預測建立在驅動共折疊過程的潛在表示之上。這種表示本質上編碼了關于生物分子相互作用的豐富信息。因此,Boltz-2在結合親和力預測方面的改進是由結構建模的進步所推動的。這些進步源于:(1)擴展訓練數據,超越靜態結構,包括實驗和分子動力學集合;(2)顯著擴大多樣模態下的蒸餾數據集;(3)通過基于實驗方法、用戶定義的距離約束和多鏈模板集成來增強用戶控制。
整體解析:
這段文字主要討論了Boltz-2在結合親和力預測方面的改進與其背后的驅動因素。首先提到的是表示學習的重要性,其中潛在表示(latent representation)是親和力預測的核心,它能夠捕捉生物分子相互作用的關鍵信息。接著指出,Boltz-2的性能提升得益于結構建模的進步,而這些進步來源于三個方面:一是擴展了訓練數據的范圍,使其不僅限于靜態結構,還包括實驗數據和分子動力學模擬數據;二是通過增加蒸餾數據集的規模和多樣性來提高模型的泛化能力;三是通過引入用戶自定義參數(如實驗方法和距離約束)以及多鏈模板集成,增強了模型的靈活性和可控性。這表明,Boltz-2不僅在算法層面有所改進,還在數據和用戶交互方面進行了優化。
Boltz-2藥物發現的重要場景
Boltz-2對親和力的準確預測,使得它可用于藥物發現的重要場景:
苗頭化合物發現(Hit discovery):該模型在高通量篩選中區分結合劑(binders)與誘餌(decoys),并在MF-PCBA基準測試中實現了顯著的富集增益 [Buterez等, 2023],其表現優于對接(docking)和機器學習(ML)方法。具體來說,該模型的任務是從高通量篩選(high-throughput screens)中識別出真正的結合劑(binders),同時排除非活性分子(decoys)。通過使用MF-PCBA基準測試對該模型進行評估,結果表明它在富集增益(enrichment gains)方面表現優異。富集增益是指模型能夠以更高的比例挑選出真正有活性的分子,相較于隨機選擇或傳統方法更具優勢。此外,該模型的表現超越了傳統的對接(docking)方法以及基于機器學習(ML)的方法,體現了其在藥物篩選中的潛力。
苗頭化合物到先導化合物以及先導化合物優化(Hit-to-lead and lead optimization): 是藥物發現和開發過程中的兩個關鍵階段。首先,“Hit-to-lead” 指的是在初期篩選中找到具有活性的化合物(稱為“hit”),然后通過進一步的化學修飾和生物學測試,將這些“hit”轉化為更具潛力和選擇性的候選分子,即“lead”。接下來,“lead optimization” 是對這些先導化合物進行優化的過程,通過對化合物的結構進行調整,提高其藥效、選擇性、代謝穩定性等特性,以使其更接近成為臨床試驗中的候選藥物。這兩個階段是藥物研發的重要步驟,為后續的臨床研究奠定基礎。
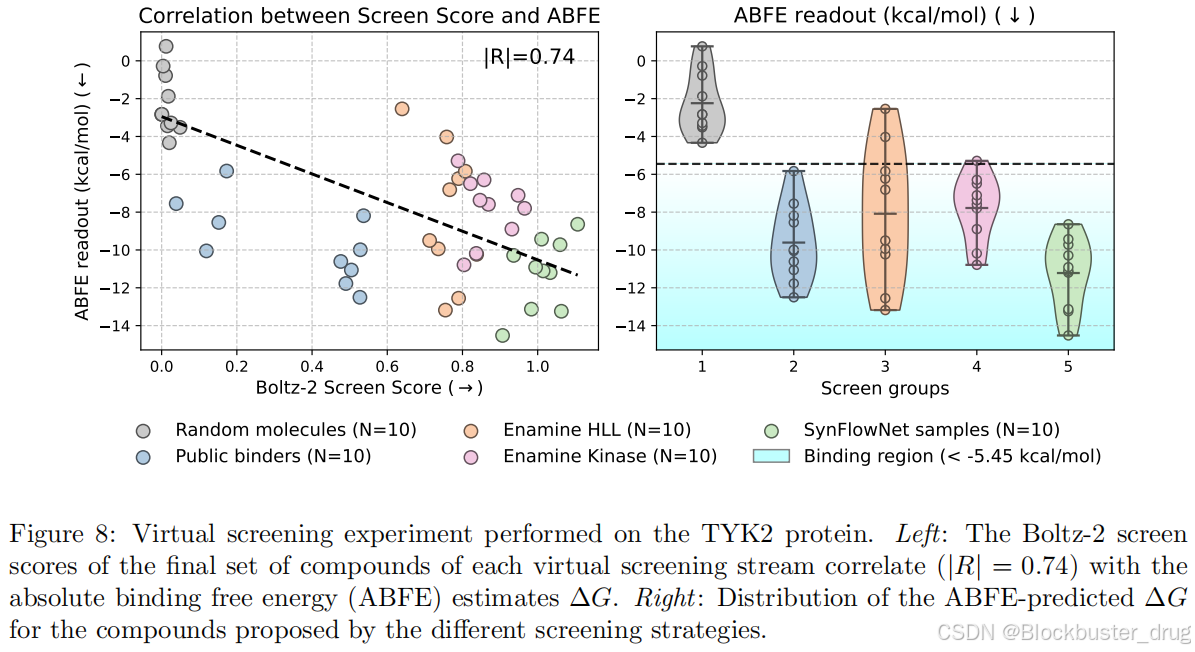
從頭生成(de-novo Generation):De-novo Generation(從頭生成)與生成模型相結合 [Cretu et al., 2024],Boltz-2 能夠發現新的結合劑。在針對 TYK2 靶點的前瞻性篩選中,該流程能夠生成多樣化、可合成的高親和力結合劑,這一點通過絕對結合自由能 (ABFE) 模擬 [Wu et al., 2025] 估算得出。這種方法特別適用于發現針對特定靶點(如 TYK2)的新結合劑(binders)。文中提到,該流程不僅能夠生成多樣化的分子,還確保這些分子具有高親和力(high-affinity),并且可以通過化學手段合成(synthetizable)。為了驗證這些分子的實際效果,研究團隊使用了絕對結合自由能(ABFE)模擬來評估分子與靶點之間的結合強度。這種結合強度是藥物設計中的關鍵指標,直接反映了分子作為潛在藥物的潛力。
Boltz-2的改進及對比優勢
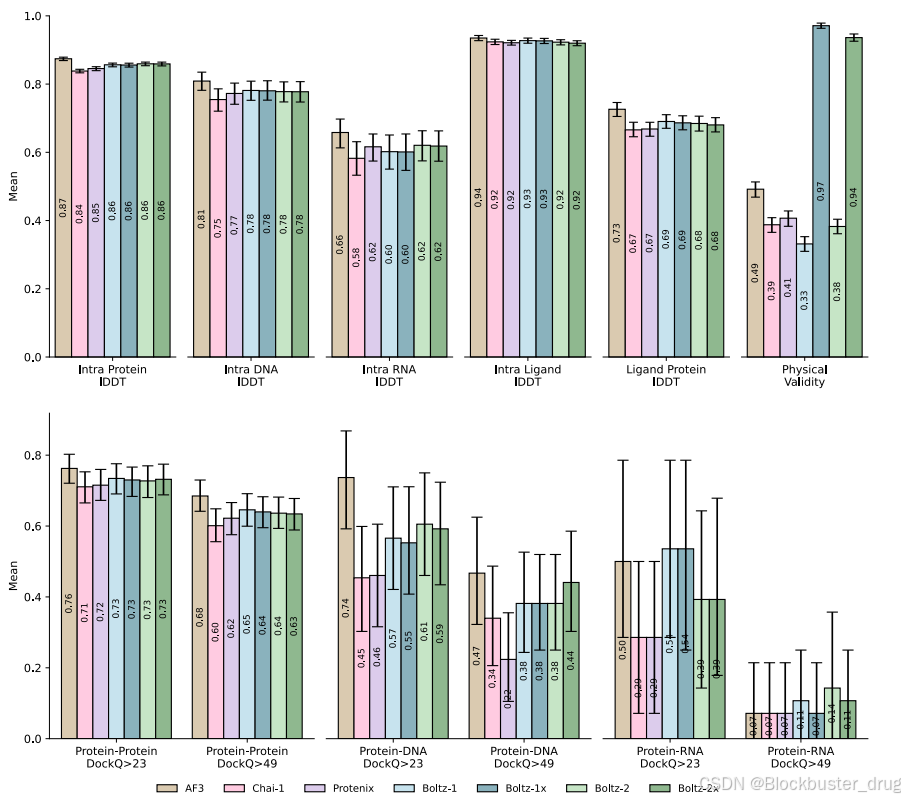
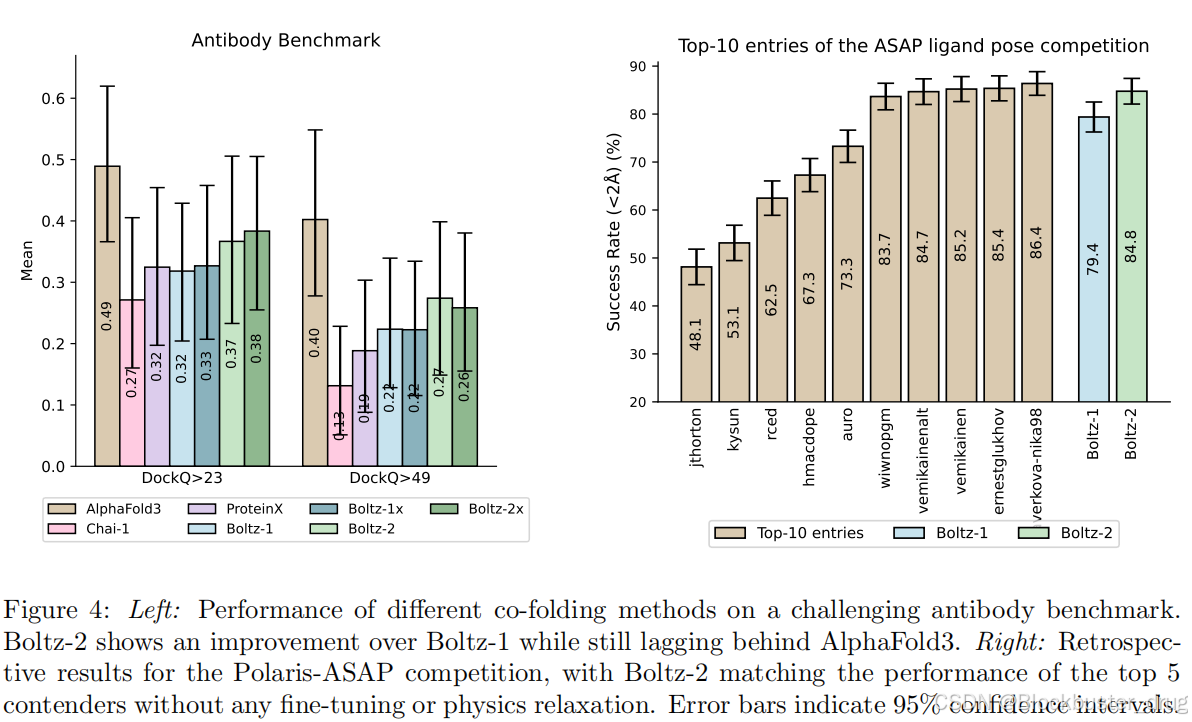
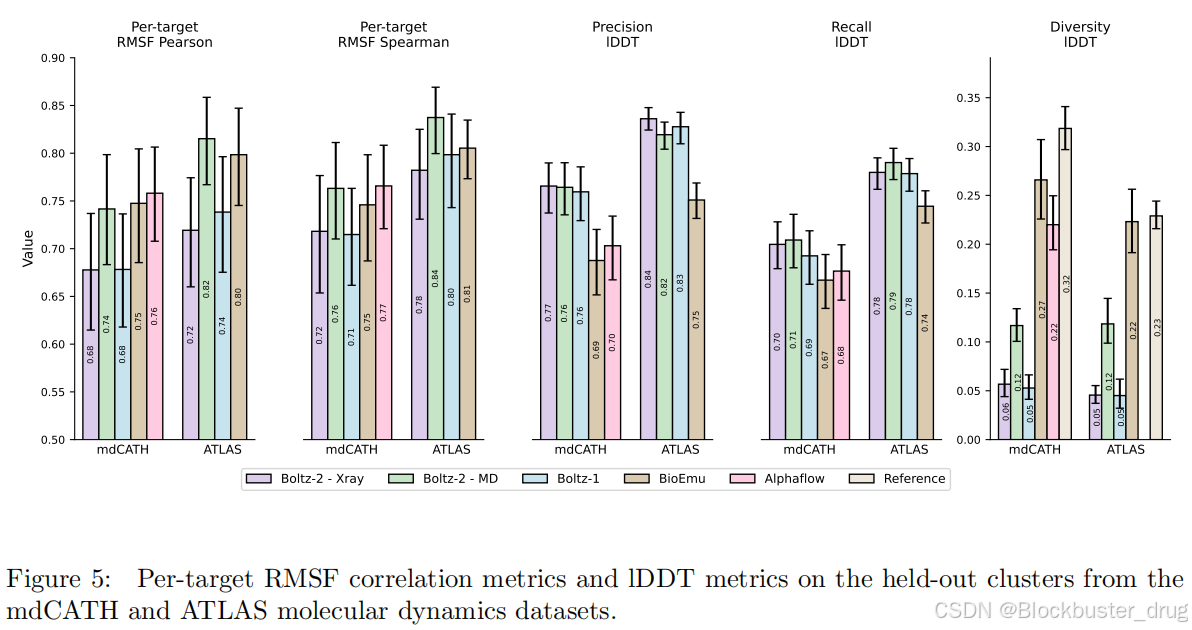
與Boltz-1相比,Boltz-2在跨模態的晶體結構預測方面有所改進,特別是在抗體-抗原復合物等具有挑戰性的目標上表現尤為突出。當與分子動力學模擬進行基準比較時,Boltz-2在預測關鍵動態特性(如均方根波動RMSF)方面的性能可以與近期的專用模型(例如AlphaFlow [Jing et al., 2024] 和 BioEmu [Lewis et al., 2025])相媲美。
2. 安裝及應用
安裝:
創建一個conda環境,boltz-2
conda create -n boltz-2 python=3.12
激活環境,后續在環境中操作。
conda activate boltz-2
方法1:創建一個新的python環境(比如使用conda),使用pip安裝:
pip install boltz -U方法2:下載并安裝最新版本:
git clone https://github.com/jwohlwend/boltz.git
cd boltz; pip install -e .使用推理:
boltz predict input_path --use_msa_serverinput_path 應指向一個 YAML 文件,或用于批量處理的 YAML 文件目錄,描述您想要建模的生物分子及其預測的屬性(例如親和力)。要查看所有可用選項:boltz predict --help;有關這些輸入格式的更多信息,請參閱我們的預測說明。默認情況下,boltz 命令將運行模型的最新版本。
YAML 格式更加靈活,允許更復雜的輸入,尤其是共價鍵相關的輸入。YAML 的架構如下:
sequences:- ENTITY_TYPE:id: CHAIN_ID sequence: SEQUENCE # only for protein, dna, rnasmiles: 'SMILES' # only for ligand, exclusive with ccdccd: CCD # only for ligand, exclusive with smilesmsa: MSA_PATH # only for proteinmodifications:- position: RES_IDX # index of residue, starting from 1ccd: CCD # CCD code of the modified residuecyclic: false- ENTITY_TYPE:id: [CHAIN_ID, CHAIN_ID] # multiple ids in case of multiple identical entities...
constraints:- bond:atom1: [CHAIN_ID, RES_IDX, ATOM_NAME]atom2: [CHAIN_ID, RES_IDX, ATOM_NAME]- pocket:binder: CHAIN_IDcontacts: [[CHAIN_ID, RES_IDX/ATOM_NAME], [CHAIN_ID, RES_IDX/ATOM_NAME]]max_distance: DIST_ANGSTROM- contact:token1: [CHAIN_ID, RES_IDX/ATOM_NAME]token2: [CHAIN_ID, RES_IDX/ATOM_NAME]max_distance: DIST_ANGSTROMtemplates:- cif: CIF_PATH # if only a path is provided, Boltz will find the best matchings- cif: CIF_PATHchain_id: CHAIN_ID # optional, specifiy which chain to find a template for- cif: CIF_PATHchain_id: [CHAIN_ID, CHAIN_ID] # can be more than onetemplate_id: [TEMPLATE_CHAIN_ID, TEMPLATE_CHAIN_ID]
properties:- affinity:binder: CHAIN_ID
sequences輸入中每個唯一的鏈/分子都有一個條目。每個聚合物實體都表示為ENTITY_TYPE?、protein或dna,rna并具有一個sequence屬性。非聚合物實體用ENTITY_TYPE等于表示ligand,并具有smiles或ccd屬性。CHAIN_ID是每個鏈/分子的唯一標識符,如果結構中存在多個相同的實體,則應將其設置為列表。對于蛋白質,msa默認情況下需要鍵,但可以通過傳遞--use_msa_server標志來省略,該標志將使用 mmseqs2 服務器自動生成 MSA。如果您希望使用預先計算的 MSA,請使用msa屬性,該屬性MSA_PATH指示包含該蛋白質 MSA 的文件的路徑。如果您希望明確運行單序列模式(通常不建議這樣做,因為它會損害模型性能),您可以使用該蛋白質的特殊關鍵字(例如: ).a3m來實現。對于自定義 MSA,您可能希望向模型指示配對鍵。您可以使用 CSV 格式(而不是 a3m)來實現此目的,其中包含兩列:包含蛋白質序列和 ,其中 是一個唯一標識符,指示每個蛋白質鏈在 CSV 文件中的匹配行。emptymsa: emptysequencekey
字段modifications為可選字段,用于指定聚合物中的修飾殘基(protein,dna或rna)。position字段指定殘基的索引(從 1 開始),ccd為修飾殘基的 CCD 代碼。此字段目前僅支持 CCD 配體。 標志cyclic應用于指定環狀聚合物鏈(而非配體)。
constraints是一個可選字段,允許您指定有關輸入結構的附加信息。
-
該
bond約束指定兩個原子之間的共價鍵(atom1和atom2)。目前僅支持CCD配體和規范殘基,CHAIN_ID指的是上面設置的殘基的id,RES_IDX是殘基的索引(從1開始)(配體為1),ATOM_NAME是標準化的原子名稱(可以在RCSB網站上該組件的CIF文件中驗證)。 -
約束
pocket指定與配體結合的殘基,其中binder表示與口袋(可以是分子、蛋白質、DNA 或 RNA)結合的鏈, 表示contacts與口袋結合的鏈和殘基索引列表(從 1 開始)。該模型目前僅支持指定單條鏈(以及其他鏈中binder任意數量的殘基)。contacts
templates是一個可選字段,允許您為預測指定結構模板。您至少必須提供結構模板的路徑,該路徑必須以 CIF 文件的形式提供。如果您希望明確定義 YAML 中的哪些鏈應該使用此 CIF 文件進行模板化,您可以使用該chain_id條目來指定它們。無論是否提供一組 ID,Boltz 都會從提供的模板中找到最佳匹配的鏈。如果您希望自己明確定義映射,可以提供相應的 template_id。請注意,只有蛋白質鏈可以進行模板化。
properties是一個可選字段,用于指定是否要計算親和力。如果啟用,您還必須提供與要計算親和力的小分子對應的 chain_id。只能指定一個分子進行親和力計算,并且該分子必須是配體鏈(不能是蛋白質、DNA 或 RNA)。
version: 1
sequences:- protein:id: [A, B]sequence: MVTPEGNVSLVDESLLVGVTDEDRAVRSAHQFYERLIGLWAPAVMEAAHELGVFAALAEAPADSGELARRLDCDARAMRVLLDALYAYDVIDRIHDTNGFRYLLSAEARECLLPGTLFSLVGKFMHDINVAWPAWRNLAEVVRHGARDTSGAESPNGIAQEDYESLVGGINFWAPPIVTTLSRKLRASGRSGDATASVLDVGCGTGLYSQLLLREFPRWTATGLDVERIATLANAQALRLGVEERFATRAGDFWRGGWGTGYDLVLFANIFHLQTPASAVRLMRHAAACLAPDGLVAVVDQIVDADREPKTPQDRFALLFAASMTNTGGGDAYTFQEYEEWFTAAGLQRIETLDTPMHRILLARRATEPSAVPEGQASENLYFQmsa: ./examples/msa/seq1.a3m- ligand:id: [C, D]ccd: SAH- ligand:id: [E, F]smiles: 'N[C@@H](Cc1ccc(O)cc1)C(=O)O'例如,要使用 10 個回收步驟和 25 個樣本(AlphaFold3 的默認參數)預測結構,請使用:
boltz predict input_path --recycling_steps 10 --diffusion_samples 25| 選項 | 類型 | 默認 | 描述 |
| --out_dir | PATH | ./ | 保存預測的路徑。 |
| --cache | PATH | ~/.boltz | 下載數據和模型的目錄。BOLTZ_CACHE如果設置,將使用環境變量作為絕對路徑 |
| --checkpoint | PATH | 沒有任何 | 可選檢查點。默認使用提供的 Boltz-2 模型。 |
| --devices | INTEGER | 1 | 用于預測的設備數量。 |
| --accelerator | [gpu,cpu,tpu] | gpu | 用于預測的加速器。 |
| --recycling_steps | INTEGER | 3 | 用于預測的回收步驟數。 |
| --sampling_steps | INTEGER | 200 | 用于預測的采樣步驟數。 |
| --diffusion_samples | INTEGER | 1 | 用于預測的擴散樣本的數量。 |
| --max_parallel_samples | INTEGER | 5 | 并行預測的最大樣本數。 |
| --step_scale | FLOAT | 1.638 | 步長與擴散過程采樣分布的溫度有關。步長越低,樣本間的多樣性越高(建議在 1 到 2 之間)。 |
| --output_format | [pdb,mmcif] | mmcif | 用于預測的輸出格式。 |
| --num_workers | INTEGER | 2 | 用于預測的數據加載器工作者的數量。 |
| --method | 字符串 | 沒有任何 | 用于預測的方法。 |
| --preprocessing-threads | INTEGER | multiprocessing.cpu_count() | 用于預處理的線程數。 |
| --affinity_mw_correction | FLAG | FALSE | 是否將分子量校正添加到親和力值頭。 |
| --sampling_steps_affinity | INTEGER | 200 | 用于親和力預測的采樣步驟數。 |
| --diffusion_samples_affinity | INTEGER | 5 | 用于親和力預測的擴散樣本數量。 |
| --affinity_checkpoint | PATH | 沒有任何 | 可選的親和性檢查點。默認使用提供的 Boltz-2 模型。 |
| --max_msa_seqs | INTEGER | 8192 | 用于預測的 MSA 序列的最大數量。 |
| --subsample_msa | FLAG | FALSE | 是否對 MSA 進行子采樣。 |
| --num_subsampled_msa | INTEGER | 1024 | 要進行子采樣的 MSA 序列的數量。 |
| --no_trifast | FLAG | FALSE | 是否不使用 trifast 內核進行三角更新。 |
| --override | FLAG | FALSE | 如果發現,是否覆蓋現有預測。 |
| --use_msa_server | FLAG | FALSE | 是否使用 msa 服務器生成 msa。 |
| --msa_server_url | 字符串 | https://api.colabfold.com | MSA 服務器 URL。僅當設置了 --use_msa_server 時使用。 |
| --msa_pairing_strategy | 字符串 | greedy | 使用的配對策略。僅當設置了 --use_msa_server 時才使用。選項包括“greedy”和“complete”。 |
| --use_potentials | FLAG | FALSE | 是否使用推理時間潛力運行原始 Boltz-2 模型。 |
| --write_full_pae | FLAG | FALSE | 是否將完整的 PAE 矩陣保存為文件。 |
| --write_full_pde | FLAG | FALSE | 是否將完整的 PDE 矩陣保存為文件。 |
親和力輸出中有兩個主要預測:affinity_pred_value 和 affinity_probability_binary。它們在截然不同的數據集上進行訓練,并采用不同的監督方法,因此應在不同的情況下使用。affinity_probability_binary 字段應用于從誘餌中檢測結合劑,例如在發現目標化合物階段。其值范圍為 0 到 1,表示預測配體為結合劑的概率。affinity_pred_value 旨在測量不同結合劑的特定親和力,以及這種親和力如何隨著分子的細微修改而變化。這應該用于配體優化階段,例如從目標化合物到先導化合物和先導化合物優化。它將結合親和力值報告為 log(IC50),該值源自以 μM 為單位測量的 IC50。有關如何運行親和力預測和解析輸出的更多詳細信息,請參閱我們的預測說明。
輸出:
out_dir/
├── lightning_logs/ # Logs generated during training or evaluation
├── predictions/ # Contains the model's predictions├── [input_file1]/├── [input_file1]_model_0.cif # The predicted structure in CIF format, with the inclusion of per token pLDDT scores├── confidence_[input_file1]_model_0.json # The confidence scores (confidence_score, ptm, iptm, ligand_iptm, protein_iptm, complex_plddt, complex_iplddt, chains_ptm, pair_chains_iptm)├── affinity_[input_file1].json # The affinity scores (affinity_pred_value, affinity_probability_binary, affinity_pred_value1, affinity_probability_binary1, affinity_pred_value2, affinity_probability_binary2)├── pae_[input_file1]_model_0.npz # The predicted PAE score for every pair of tokens├── pde_[input_file1]_model_0.npz # The predicted PDE score for every pair of tokens├── plddt_[input_file1]_model_0.npz # The predicted pLDDT score for every token...└── [input_file1]_model_[diffusion_samples-1].cif # The predicted structure in CIF format...└── [input_file2]/...
└── processed/ # Processed data used during execution 該predictions文件夾為每個輸入文件包含一個唯一的文件夾。輸入文件夾包含diffusion_samples按置信度分數排序的 output_format 格式的預測結果,以及包含置信度模型和親和度模型預測結果的附加文件。該processed文件夾包含模型在推理過程中使用的已處理輸入文件。
輸出置信度.json文件包含特定樣本的各種聚合置信度得分。文件結構如下:
{"confidence_score": 0.8367, # Aggregated score used to sort the predictions, corresponds to 0.8 * complex_plddt + 0.2 * iptm (ptm for single chains)"ptm": 0.8425, # Predicted TM score for the complex"iptm": 0.8225, # Predicted TM score when aggregating at the interfaces"ligand_iptm": 0.0, # ipTM but only aggregating at protein-ligand interfaces"protein_iptm": 0.8225, # ipTM but only aggregating at protein-protein interfaces"complex_plddt": 0.8402, # Average pLDDT score for the complex"complex_iplddt": 0.8241, # Average pLDDT score when upweighting interface tokens"complex_pde": 0.8912, # Average PDE score for the complex"complex_ipde": 5.1650, # Average PDE score when aggregating at interfaces "chains_ptm": { # Predicted TM score within each chain"0": 0.8533,"1": 0.8330},"pair_chains_iptm": { # Predicted (interface) TM score between each pair of chains"0": {"0": 0.8533,"1": 0.8090},"1": {"0": 0.8225,"1": 0.8330}}
}confidence_score,ptm分數plddt(及其界面和單個鏈類似物)的范圍是[0, 1],其中值越高表示置信度越高。pde分數的單位是埃,其中值越低表示置信度越高。
輸出親和性.json文件的組織如下:
{"affinity_pred_value": 0.8367, # Predicted binding affinity from the enseble model"affinity_probability_binary": 0.8425, # Predicted binding likelihood from the ensemble model"affinity_pred_value1": 0.8225, # Predicted binding affinity from the first model of the ensemble"affinity_probability_binary1": 0.0, # Predicted binding likelihood from the first model in the ensemble"affinity_pred_value2": 0.8225, # Predicted binding affinity from the second model of the ensemble"affinity_probability_binary2": 0.8402, # Predicted binding likelihood from the second model in the ensemble
}親和力輸出中有兩個主要預測:affinity_pred_value和affinity_probability_binary。它們在截然不同的數據集上進行訓練,并采用不同的監督方法,因此應該在不同的情境中使用。添加評論更多操作
該affinity_probability_binary字段應用于檢測結合物和誘餌,例如在發現目標物階段。其值范圍為 0 到 1,表示預測配體為結合物的概率。
旨在affinity_pred_value測量不同結合劑的特異性親和力,以及其如何隨著分子的微小修改而變化。這應該用于配體優化階段,例如命中到先導化合物和先導化合物優化。它報告的結合親和力值為log(IC50),源自于IC50測量的μM。值越低,預測的結合力越強,例如:
- IC50 10?9M?我們的模型輸出 ?3(強親和力分子)
- IC50 10?6M?我們的模型輸出 0(中等親和力分子)
- IC50 10?4M?我們的模型輸出 2(弱親和力)
kcal/mol您可以使用y --> (6 - y) * 1.364模型y的預測將模型的輸出轉換為 pIC50 。
3. 使用示例
下載boltz程序包:
git clone https://github.com/jwohlwend/boltz.git使用examples中的affinity計算實例:
cd boltzboltz predict ./examples/affinity.yaml --use_msa_server初次運行,會下載解壓CCD data,在/home/user/.boltz/mols目錄下,大小1.8G,包含45227個mol文件。
然后會下載?Boltz-2 weights,在?/home/user/.boltz目錄下,名稱為boltz2_conf.ckpt,大小2.3G。
affinity預測,首次會下載affinity weights在/home/user/.boltz目錄下,名稱boltz2_aff.ckpt,大小2.1G。
錯誤提示1:RuntimeError: PytorchStreamReader failed
原因1:主要是weights文件不完整,可以提前下載放在/home/user/.boltz下。下載URL在/boltz/src/boltz/main.py文件中,如下:
CCD_URL = "https://huggingface.co/boltz-community/boltz-1/resolve/main/ccd.pkl"
MOL_URL = "https://huggingface.co/boltz-community/boltz-2/resolve/main/mols.tar"
BOLTZ1_URL_WITH_FALLBACK = [
? ? "https://model-gateway.boltz.bio/boltz1_conf.ckpt",
? ? "https://huggingface.co/boltz-community/boltz-1/resolve/main/boltz1_conf.ckpt",
]
BOLTZ2_URL_WITH_FALLBACK = [
? ? "https://model-gateway.boltz.bio/boltz2_conf.ckpt",
? ? "https://huggingface.co/boltz-community/boltz-2/resolve/main/boltz2_conf.ckpt",
]
BOLTZ2_AFFINITY_URL_WITH_FALLBACK = [
? ? "https://model-gateway.boltz.bio/boltz2_aff.ckpt",
? ? "https://huggingface.co/boltz-community/boltz-2/resolve/main/boltz2_aff.ckpt",
]
結果文件夾名稱為:boltz_results_affinity,內容如下:
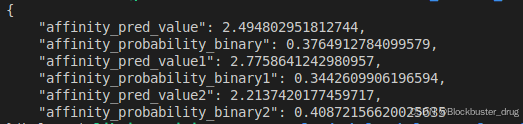
結果在affinity_affinity.json文件中,可以看到,親和力以log(IC50)為單位,等于6-2.5=3.5,大致等于300μM,是一個弱的binder。
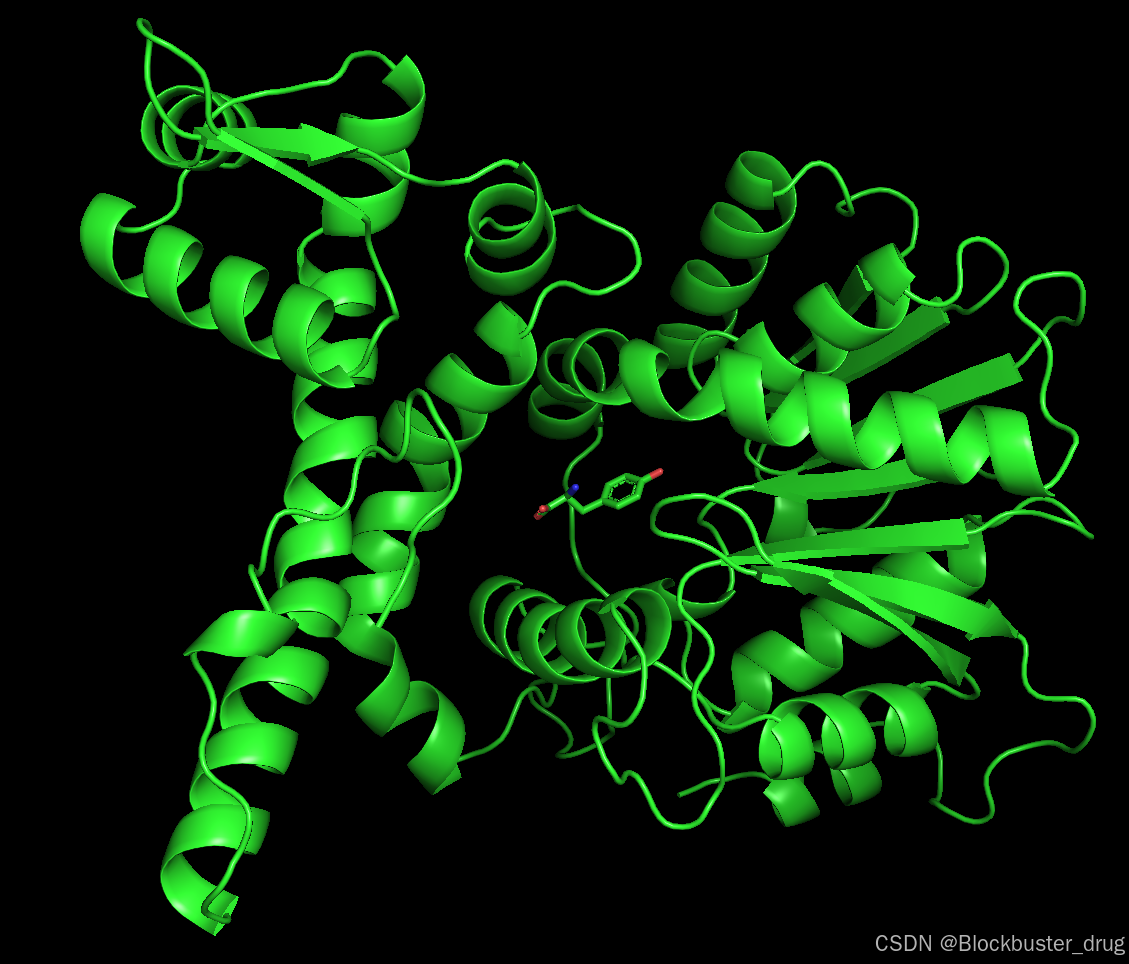
復合物結構文件為affinity_model_0.cif,使用pymol打開:
pymol /boltz_results_affinity/predictions/affinity/affinity_model_0.cif結構如下:
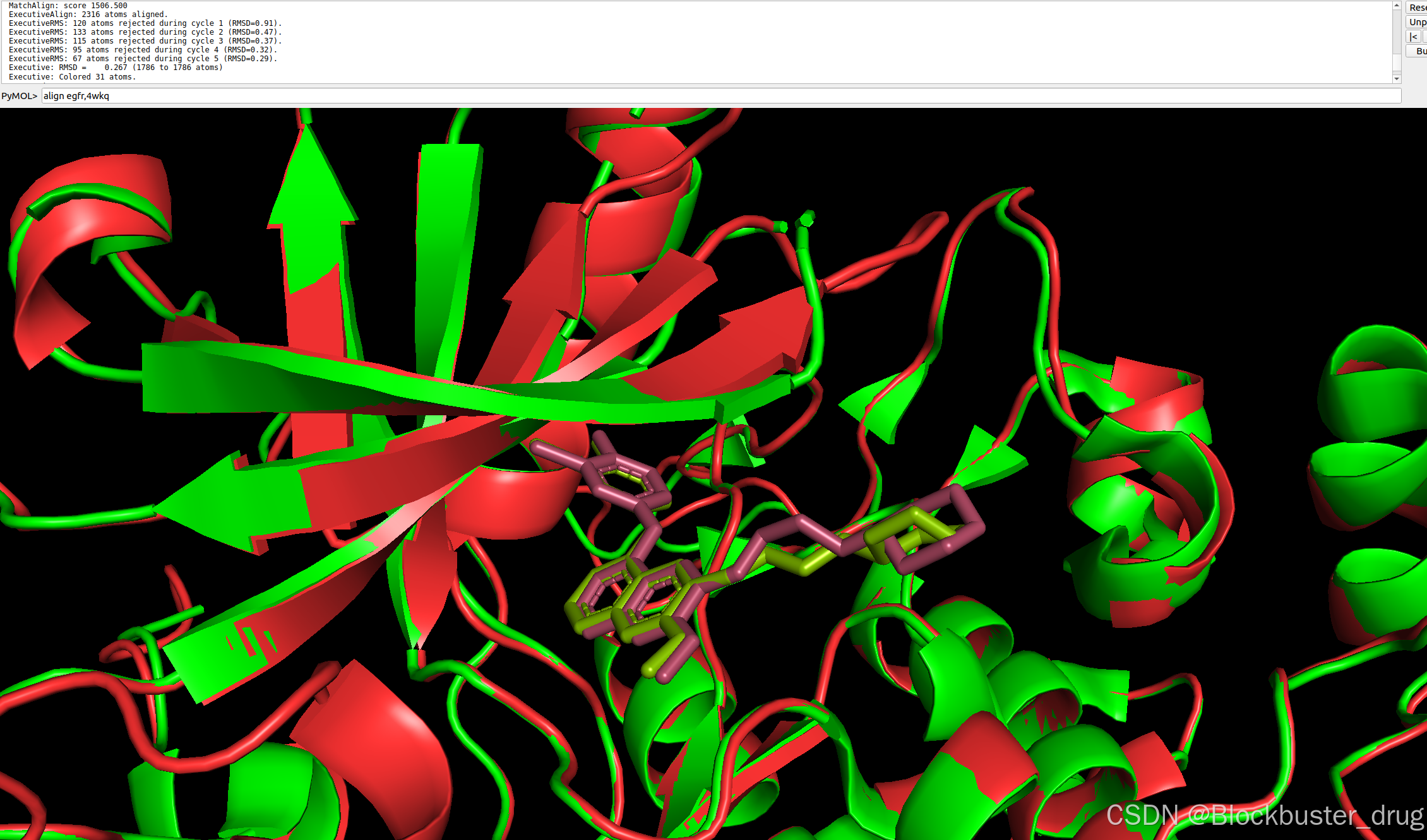
又一例,如下是EGFR抑制劑吉非替尼的親和力計算文件:
version: 1 # Optional, defaults to 1
sequences:- protein:id: Asequence: GAMGEAPNQALLRILKETEFKKIKVLGSGAFGTVYKGLWIPEGEKVKIPVAIKELREATSPKANKEILDEAYVMASVDNPHVCRLLGICLTSTVQLITQLMPFGCLLDYVREHKDNIGSQYLLNWCVQIAKGMNYLEDRRLVHRDLAARNVLVKTPQHVKITDFGLAKLLGAEEKEYHAEGGKVPIKWMALESILHRIYTHQSDVWSYGVTVWELMTFGSKPYDGIPASEISSILEKGERLPQPPICTIDVYMIMVKCWMIDADSRPKFRELIIEFSKMARDPQRYLVIQGDERMHLPSPTDSNFYRALMDEEDMDDVVDADEYLIPQQG- ligand:id: Bsmiles: 'Clc1c(F)ccc(Nc2ncnc3c2cc(OCCCN2CCOCC2)c(OC)c3)c1'
properties:- affinity:binder: B預測結果顯示,預測的復合物結構與吉非替尼-EGFR復合物的PDB結構(PDBID: 4WKQ)非常接近,RMSD=0.267。
吉非替尼的親和力為-6.56 log(IC50),低于1 μM,屬于比較強的結合,與實際情況符合。
以上是boltz-2的初步使用介紹。
參考文獻:
https://github.com/jwohlwend/boltz/tree/main/docs
 集成阿里云短信驗證碼)





策略模式:思維鏈CoT和ReAct)



技術詳解)




![[NLP]UPF基本語法及其在 native low power verification中的典型流程](http://pic.xiahunao.cn/[NLP]UPF基本語法及其在 native low power verification中的典型流程)



