一、引言
?????? microeco包是福建農林大學姚敏杰教授團隊開發的擴增子測序集成分析。該包綜合了擴增子測序下游分析的多種功能包括群落組成、多樣性、網絡分析、零模型等等。通過簡單的幾行代碼可實現復雜的分析。因此,microeco包發表以來被學界廣泛關注,截止2024年10月13日已經被引644次,是領域內高被引文章。本系列推送將持續推出microeco包分析流程,這一期是微生物網絡分析(network)。
?????? 在微生物群落生態學研究中,微生物群落之間的相互作用是揭示微生物生態系統復雜性的關鍵。微生物之間的協作、競爭等關系構成了一個復雜的生態網絡,通過這些網絡,我們能夠深入理解微生物如何在生態系統中相互影響、協同工作甚至爭奪資源。網絡分析的方法多種多樣,如下闡述了選擇microeco進行網絡分析的理由。
1.1 為什么選擇 microeco 進行網絡分析?
高效、簡便的網絡構建:僅需幾行代碼便可完成復雜的網絡構建。
支持多種相關性算法:根據研究需求,選擇不同的相關性測量方式。
強大的可視化功能:與ggplot2、igraph無縫集成,生成美觀且信息豐富的網絡圖。
拓展功能豐富:不僅可以構建網絡,還可以進行模塊性、中心性等分析,深度挖掘微生物間的相互作用。
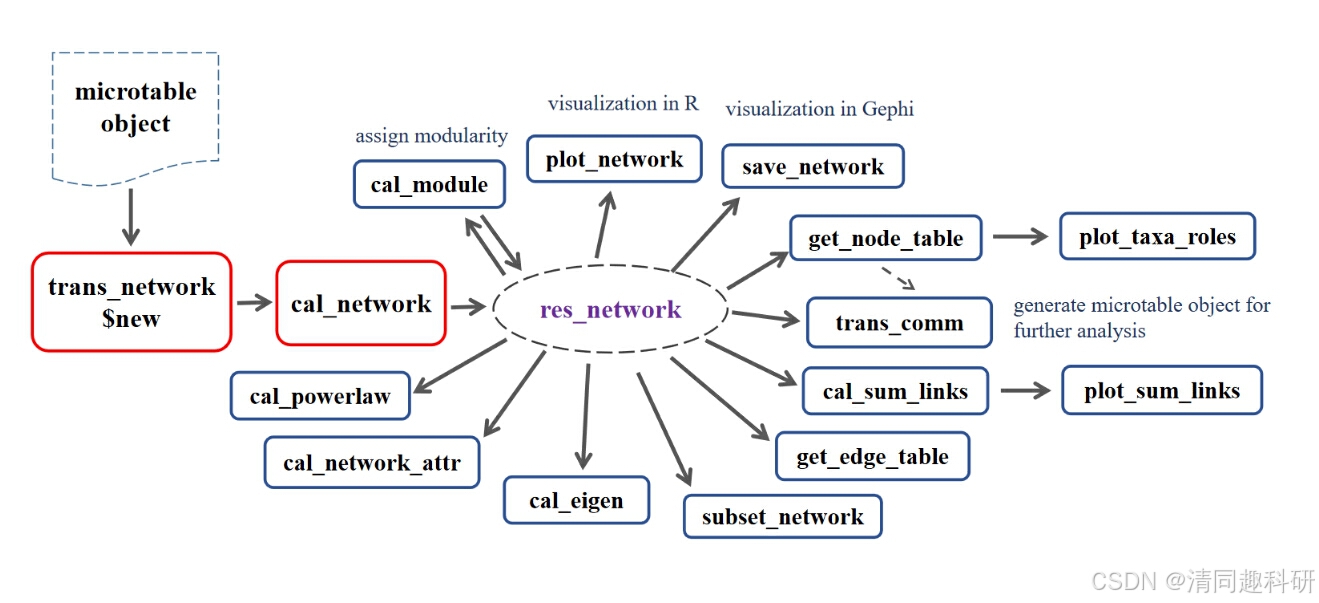
?????? 如下所示闡述了microeco包中進行微生物網絡構建和分析的流程。比較核心的內容是輸入文件的準備,也就是微表(mirotable),microeco包進行多種分析的前提也是準備好基礎數據,包括樣本信息表:sample_info,特征表otu_table,分類信息表taxonomy_table,系統發育樹phylo_tree和環境因子表env_data。當然不同的分析所需要的基礎數據也不一樣,系統發育樹和環境因子表并不是所有分析都需要的。微表準備好了可以通過幾行簡單的代碼進行下游分析。
二、文獻美圖欣賞
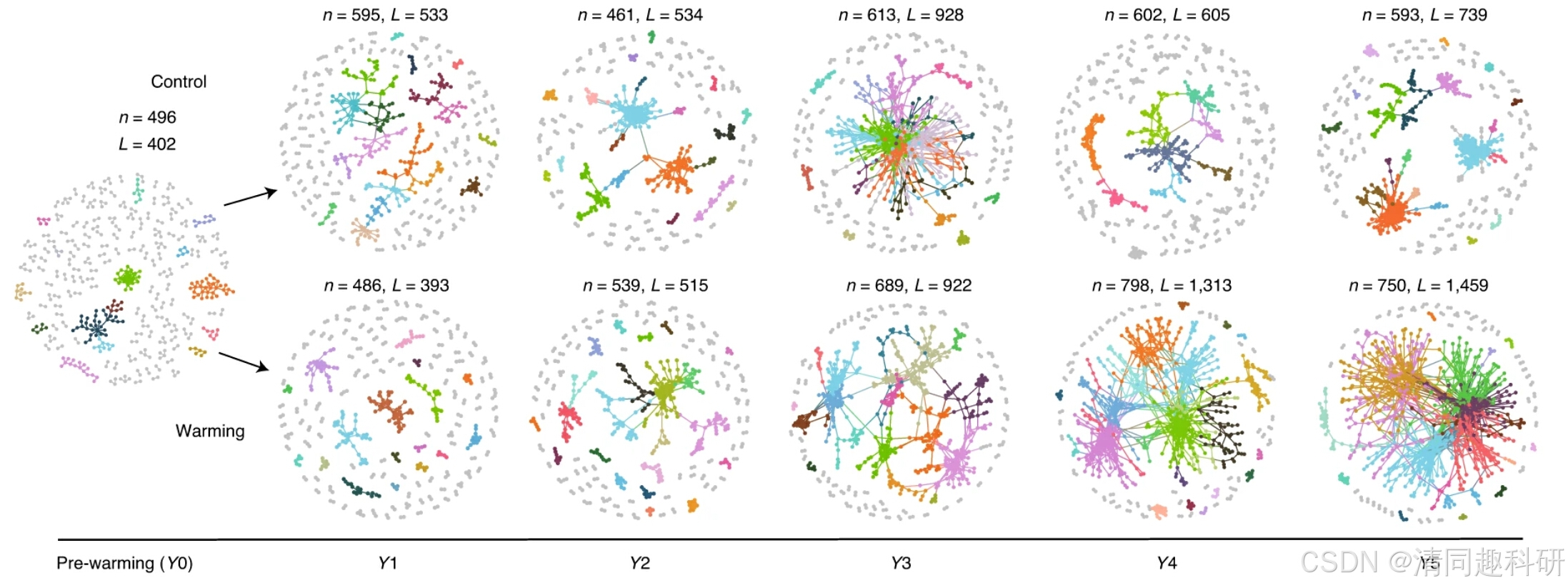
圖源:Yuan, M.M., Guo, X., Wu, L. et al.Nat. Clim. Chang. 11, 343–348 (2021).
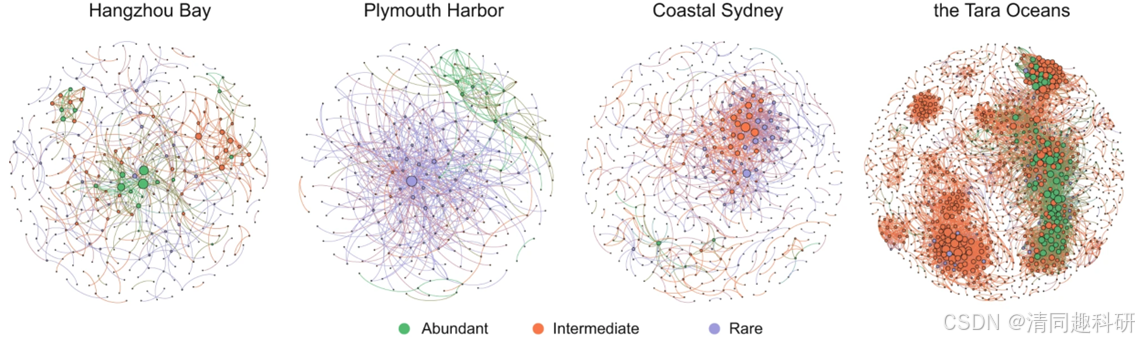
圖片來源:Dai, T., Wen, D., Bates, C.T. et al. Nat Commun 13, 175 (2022).
三、示例數據和R代碼
?????? 這套代碼基于microeco包自帶的數據進行分析,同時我們也提供了讀取自己數據的方法,可私信小編獲取代碼。
🌟數據基本操作之建立微表
#安裝microeco包
if(!require("microeco")){install.packages("microeco")}
library(microeco)
#安裝并加載ape包
if(!require("ape")){install.packages("ape")}
library(ape)##################數據基本操作#######################
# 樣本信息表;data.frame
data(sample_info_16S)
# 特征表;data.frame
data(otu_table_16S)
# 分類分配表;data.frame
data(taxonomy_table_16S)
# 系統發育樹;不是必需的;用于系統發育分析
# Newick格式;使用ape包的read.tree函數讀取樹文件
data(phylo_tree_16S)
# 加載環境數據表(如果它不在樣本表中)
data(env_data_16S)
# 使用magrittr包中的管道操作符
if(!require("magrittr")){install.packages("mgrittr")}
library(magrittr)
# 固定隨機數生成,以使結果可重復
set.seed(123)
# 使繪圖背景與教程相同
if(!require("ggplot2")){install.packages("ggplot2")}
library(ggplot2)
theme_set(theme_bw())# 統一分類信息,自動統一分類前綴
taxonomy_table_16S %<>% tidy_taxonomy# 在R6類中,'$new' 是用于創建類的新對象的原始方法
# 如果你只提供豐度表,該類可以幫助你創建一個樣本信息表
mt <- microtable$new(otu_table = otu_table_16S)
# 通常情況下添加元數據
mt <- microtable$new(otu_table = otu_table_16S, sample_table = sample_info_16S)
mt
# 讓我們創建一個包含更多信息的 microtable 對象
mt <- microtable$new(sample_table = sample_info_16S, otu_table = otu_table_16S, tax_table = taxonomy_table_16S, phylo_tree = phylo_tree_16S)
🌟數據清洗
#刪除非細菌和古菌的OTU
# 使用 R 的 subset 函數過濾 tax_table 中的分類單元
mt$tax_table %<>% base::subset(Kingdom == "k__Archaea" | Kingdom == "k__Bacteria")
mt
# 使用 grepl 函數的另一種方式
#mt$tax_table %<>% .[grepl("Bacteria|Archaea", .$Kingdom), ]#刪除線粒體或葉綠體OTU
# 這將刪除 tax_table 中包含這些詞的行,忽略分類級別并忽略大小寫。
# 因此,如果你想過濾不被認為是污染的某些分類單元,請像之前那樣使用 subset 來過濾 tax_table。
mt$filter_pollution(taxa = c("mitochondria", "chloroplast"))#使所有文件中OTU和樣本信息一致,使用tidy_dataset修剪函數
mt$tidy_dataset()
mt#使用 sample_sums() 來檢查每個樣本中的序列號
mt$sample_sums() %>% range#樣本稀疏化,每個樣本的序列數標準化為相同的數量
mt$rarefy_samples(sample.size = 10000)
mt#save_table可以將微表對象中的所有基礎數據保存到本地文件中,包括特征豐度、元數據、分類表、系統發育樹和代表序列
mt$save_table(dirpath = "basic_files", sep = ",")
🌟microeco網絡分析,構建網絡并輸出Gephi數據進行網絡繪圖
##################microeco網絡分析###################
#計算相關性矩陣
#使用WGCNA包進行計算,該包提升了計算效率,適合大規模數據
#加載并安裝WGCNA包
if(!require("WGCNA")) install.packages("WGCNA", repos = BiocManager::repositories())
#使用WGCNA進行相關性矩陣計算
t1 <- trans_network$new(dataset = mt, # 輸入數據集 mtcor_method = "spearman", # 選擇 Spearman 相關性計算方法use_WGCNA_pearson_spearman = TRUE, # 使用 WGCNA 包來計算相關性filter_thres = 0.0001 # 設置過濾閾值為 0.0001
)# 構建相關性網絡
#安裝并加載igraph包
if(!require("igraph")){install.packages("igraph")}
# 使用 p 值閾值和網絡優化進行網絡構建
#t1$cal_network(
# COR_p_thres = 0.01, # 設定相關性 p 值的閾值,只有 p 值小于 0.01 的相關性才會用于構建網絡
# COR_optimization = TRUE # 啟用網絡優化,使用算法構建更稀疏的網絡,提高網絡可解釋性
#)# 使用自定義的相關系數閾值來構建網絡(可選)
t1$cal_network(COR_p_thres = 0.01, # 設定相關性 p 值的閾值,過濾掉不顯著的相關性COR_cut = 0.7 # 使用相關系數閾值 0.7,只有相關系數大于 0.7 的變量對會被保留在網絡中
)# 對無向網絡應用 igraph 的 cluster_fast_greedy 函數進行模塊劃分
t1$cal_module(method = "cluster_fast_greedy") # 調用模塊劃分方法#將網絡保存為gexf文件,該文件可直接用于Gephi軟件進行可視化
#安裝并加載rgexf包,xfun包兼容問題,建議先安裝xfun
install.packages("xfun")
if(!require(rgexf)){install.packages('rgexf')}
#保存網絡
t1$save_network(filepath = "network.gexf")# 計算網絡屬性
t1$cal_network_attr()
t1$res_network_attr#函數get_node_table、get_edge_table和get_adjacency_matrix分別用于從網絡獲取節點屬性表、邊屬性表和鄰接矩陣。
# 獲得節點屬性
t1$get_node_table(node_roles = TRUE)
# 獲得邊屬性表
t1$get_edge_table()
# 獲得鄰接矩陣
t1$get_adjacency_matrix()#輸出節點和邊數據文件,可用于cytoscope軟件繪制網絡圖
write.csv(t1$res_edge_table,"res_edge_table.csv")
write.csv(t1$res_node_table,"res_node_table.csv")
🌟Gephi繪圖結果

🌟microeco直接繪圖和zi-pi分析,鑒別關鍵物種
#繪制網絡圖
t1$plot_network(method = "ggraph", node_color = "module",node_label = NA)#可選其他的繪圖方式
#t1$plot_network(method = "networkD3", node_color = "module")
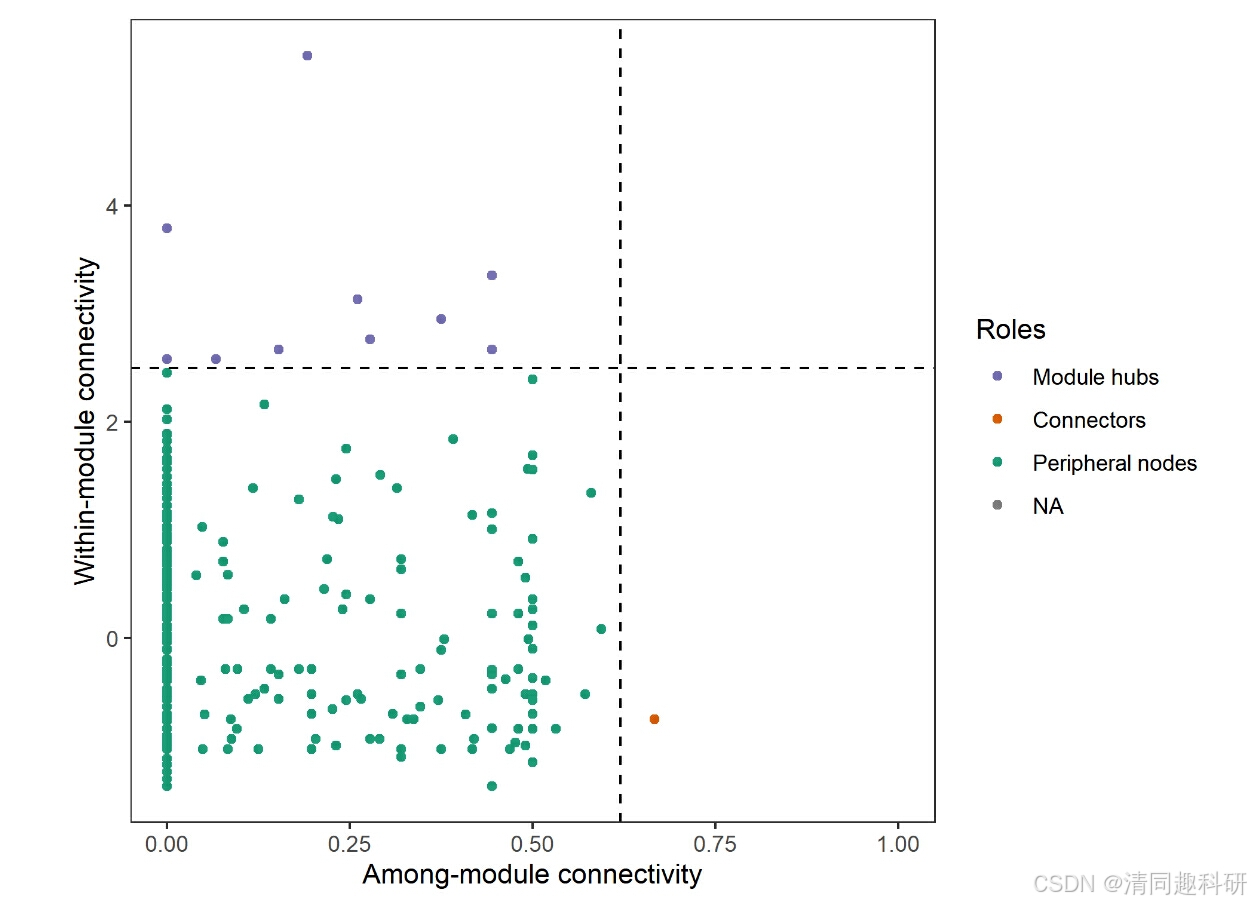
#t1$plot_network(method = "igraph")#模塊內連接和模塊間連接繪制節點分類
#add_label = TRUE 可以用于直接為點添加文本標簽
t1$plot_taxa_roles(use_type = 1)# plot node roles with phylum information
t1$plot_taxa_roles(use_type = 2)
🌟提取網絡繪制子網絡圖
從網絡中提取出來的一個部分,用于聚焦特定物種群或相互作用的分析。比如分類層級、功能屬性
#提取子網絡
#提取特定模塊的子網絡
#t1$subset_network(node = t1$res_node_table %>% base::subset(module == "M1") %>% rownames, rm_single = TRUE)
#提取特定類型邊的子網絡(正相關)
#t1$subset_network(edge = "+")#實例
# 提取樣本 'S1' 的子網絡
sub1 <- t1$subset_network(node = mt$otu_table %>% .[.[, "S1"] != 0, ] %>% rownames, rm_single = TRUE)
# 通過直接賦值創建 t2 對象
t2 <- t1
t2$res_network <- sub1 # 將提取的 'S1' 子網絡賦值給 t2 的網絡屬性# 現在 t2 包含 'S1' 的網絡,可以用于進一步分析
t2$cal_module() # 計算 t2 網絡的模塊
t2$save_network("S1.gexf") # 保存 'S1' 的網絡為 .gexf 格式文件#繪制子網絡圖
t1$plot_network(method = "ggraph", node_color = "module",node_label = NA)
#對于微生物網絡圖的繪制多種方法,可以綜合其他方法的選擇進行選取

四、參考文獻
[1] Chi Liu, Yaoming Cui, Xiangzhen Li, Minjie Yao, microeco: an R package for data mining in microbial community ecology, FEMS Microbiology Ecology, Volume 97, Issue 2, February 2021, fiaa255
[2] Dai, T., Wen, D., Bates, C.T. et al. Nutrient supply controls the linkage between species abundance and ecological interactions in marine bacterial communities. Nat Commun 13, 175 (2022).
[3] Yuan, M.M., Guo, X., Wu, L. et al. Climate warming enhances microbial network complexity and stability. Nat. Clim. Chang. 11, 343–348 (2021).
五、相關信息
!!!本文內容由小編總結互聯網和文獻內容總結整理,如若侵權,聯系立即刪除!
?!!!有需要的小伙伴評論區獲取今天的測試代碼和實例數據。
?📌示例代碼中提供了數據和代碼,小編已經測試,可直接運行。
以上就是本節所有內容。
如果這篇文章對您有用,請幫忙一鍵三連(點贊、收藏、評論、分享),讓該文章幫助到更多的小伙伴。
?
?


)




函數)
)




)


)


)