最近休息時閱讀了一本書:

在書本第5章結構信息學章節的末尾,看到了一個練習題,張貼如下:

這里作者提到了一個R包,
看著挺有意思的,所以就決定小學一下,畢竟這年頭搞分子動力學起碼是python重火力起步,看到R也能搞的屬少數。
官網參考:http://thegrantlab.org/bio3d/
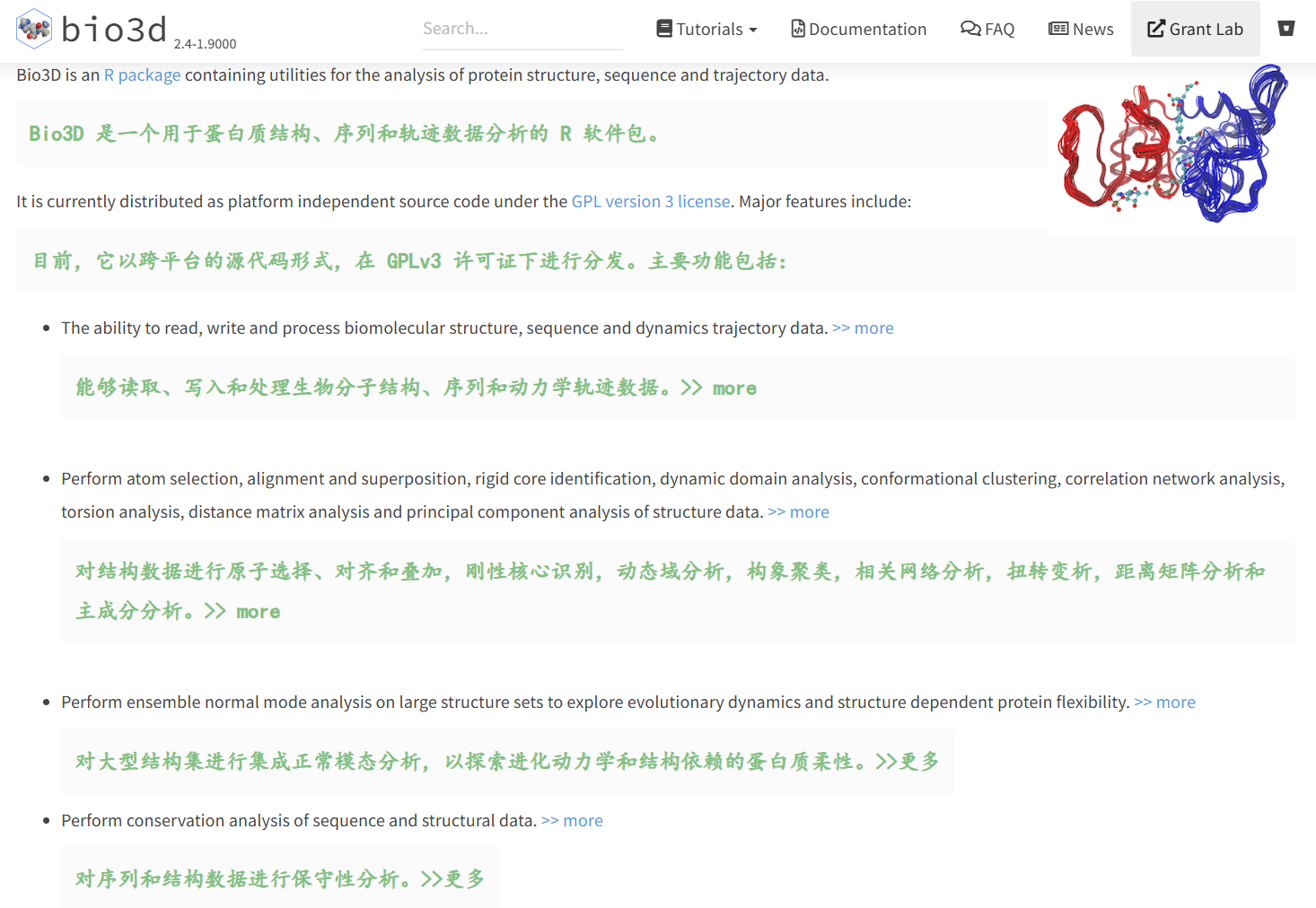

此處貼一個基本的介紹:
Bio3D是一個用于結構生物信息學分析的R語言包,主要用于生物大分子(如蛋白質和核酸)三維結構數據的處理和分析。它能夠執行各種結構分析任務,如結構對接、同源建模、分子動力學模擬軌跡分析、結構比對等。
Bio3D的核心功能包括從PDB數據庫中提取結構數據、計算結構相似性、生成結構比對和聚類分析,以及分析蛋白質動力學特征,如主成分分析(PCA)和正規模式分析(NMA)。
該包廣泛應用于蛋白質功能研究、分子演化研究以及藥物設計等領域,幫助研究者深入理解生物分子結構與功能的關系。
比如說我用python工具常做的分子動力學分析模塊——
DCCM(Dynamic cross-correlation matrices, DCCM,動態相關性矩陣),表示蛋白質中每個氨基酸的特定原子,比如說Cα原子和其他氨基酸的Cα原子之間的相關性,提供蛋白質在大尺度范圍內相關運動的一些信息。DCCM計算數值的取值范圍從完全負相關的-1.0到完全正相關的+1.0。越接近數值1表示相關性越強,正負表示兩個原子運動方向相同(反)。
計算時需要兩個文件.dcd和.pdb,
前者是蛋白質的軌跡文件,可以使用VMD進行保存,一般是分子動力學模擬之后的后續分析。我一般用它只分析分子動力學模擬穩定后的軌跡,不一定是全部軌跡;
后者是軌跡配套的蛋白質文件,選定幀數之后單獨保存為pdb格式即為蛋白質文件。
這個也能夠使用這個R包分析。
1,6QNX分析示例
參考https://www.rcsb.org/structure/6QNX
是一個SA2/SCC1/CTCF complex復合物,X射線晶體衍射獲取的結構數據。
一共是3個亞基:
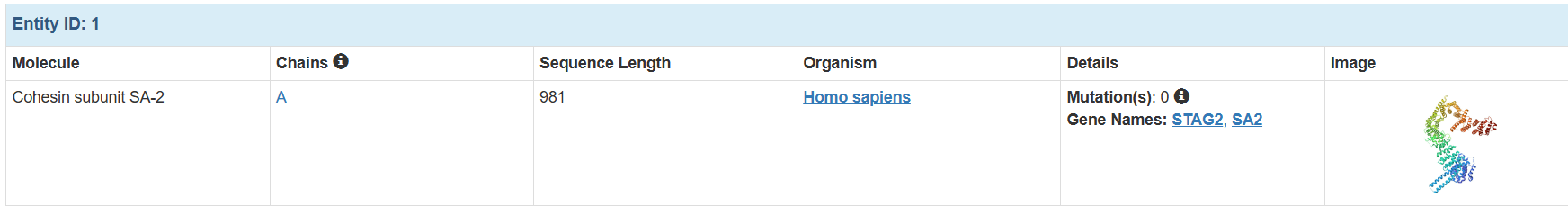
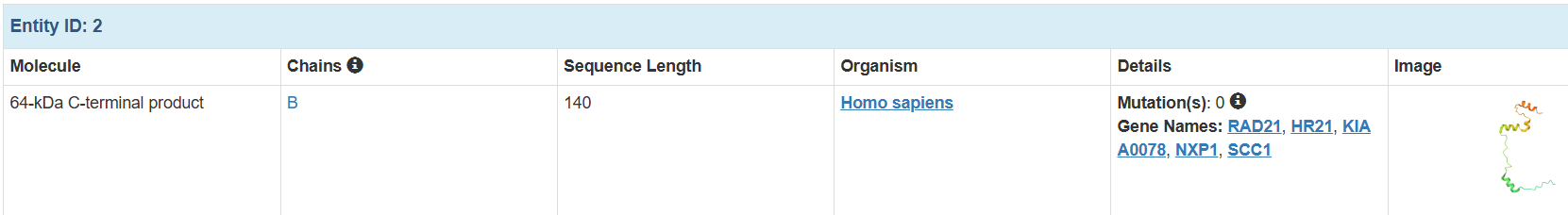
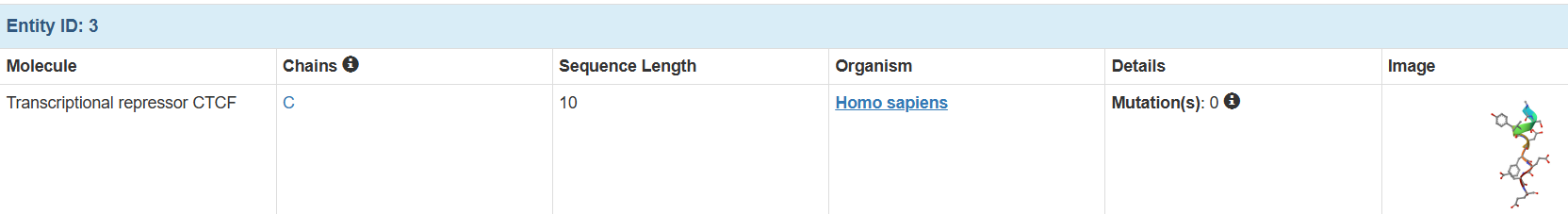
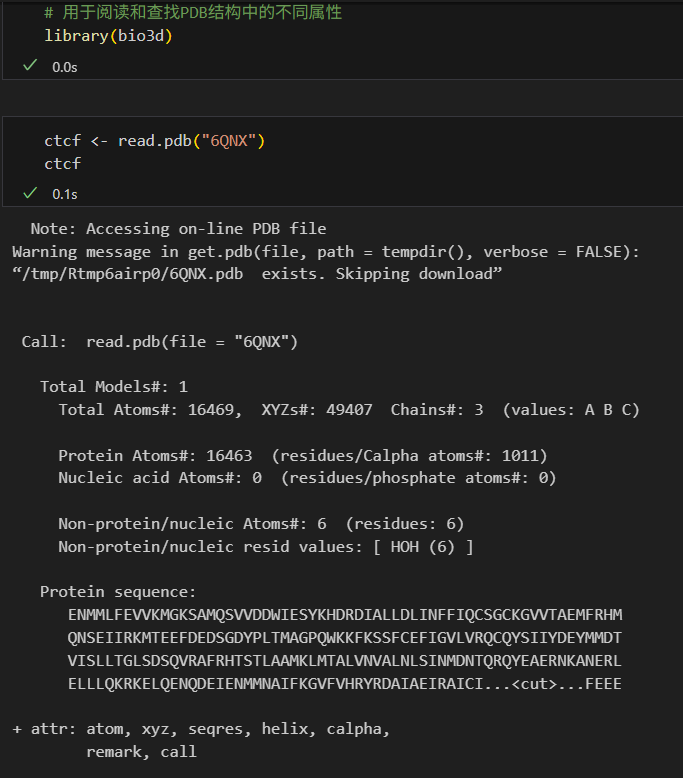
可以知道這個蛋白復合物,是由1011個氨基酸殘基,16469個原子組成的三鏈復合物。
如果想要詳細查看原子構成以及坐標信息,
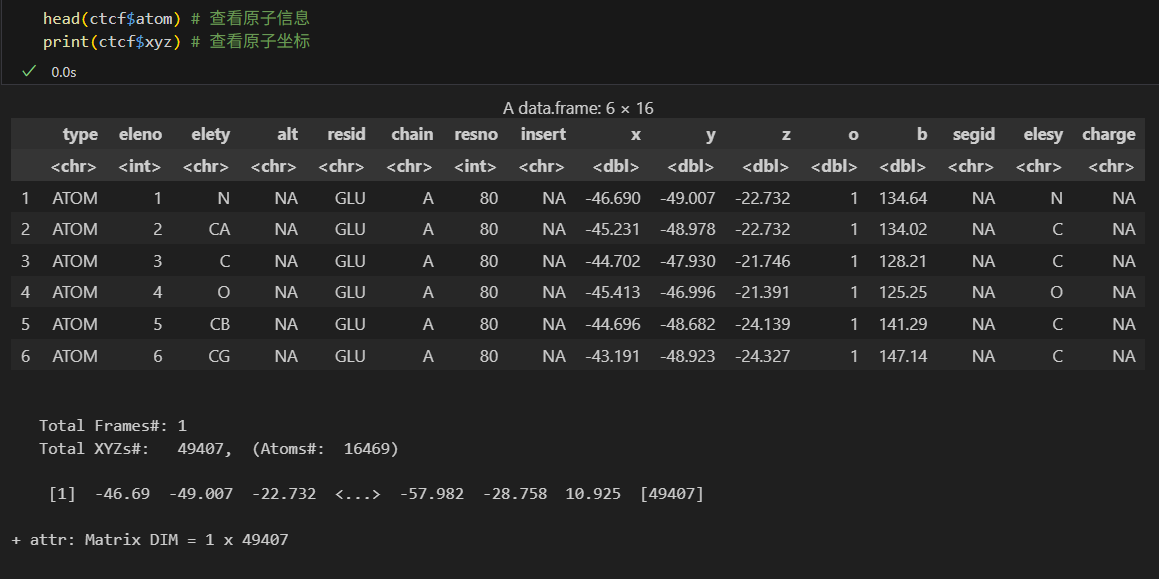
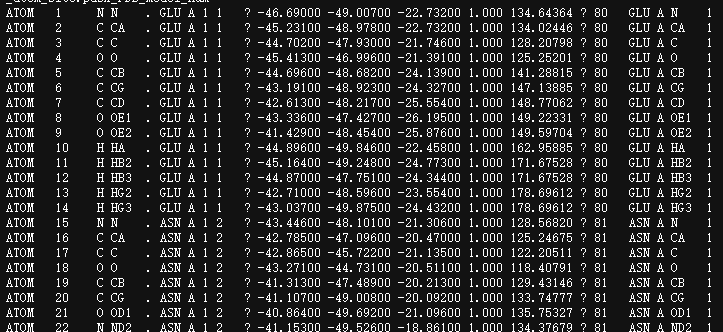
這些數據基本上都可以由mmcif文件中的坐標輔助佐證:
三維結構分析方面,因為api函數模塊很多,都值得仔細研究以及閱讀底層源碼,
這里只介紹nma()函數,也就是正規模分析(NMA,Normal Mode Analysis),從而預測示例蛋白質的柔性。
參考:http://thegrantlab.org/bio3d/reference/nma.html

詳細分析,有一個demo,可以參考:http://thegrantlab.org/bio3d_v2/tutorials/normal-mode-analysis
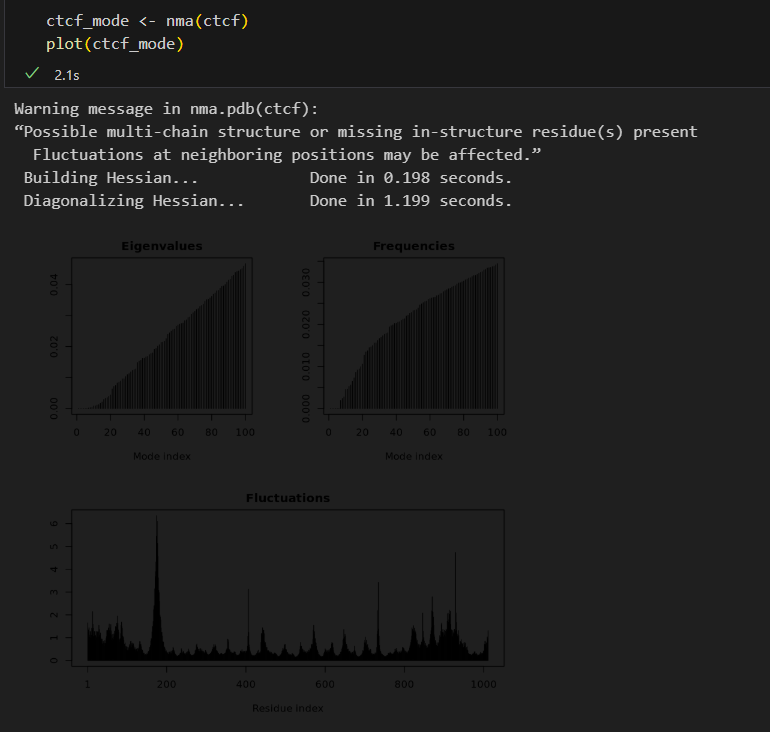
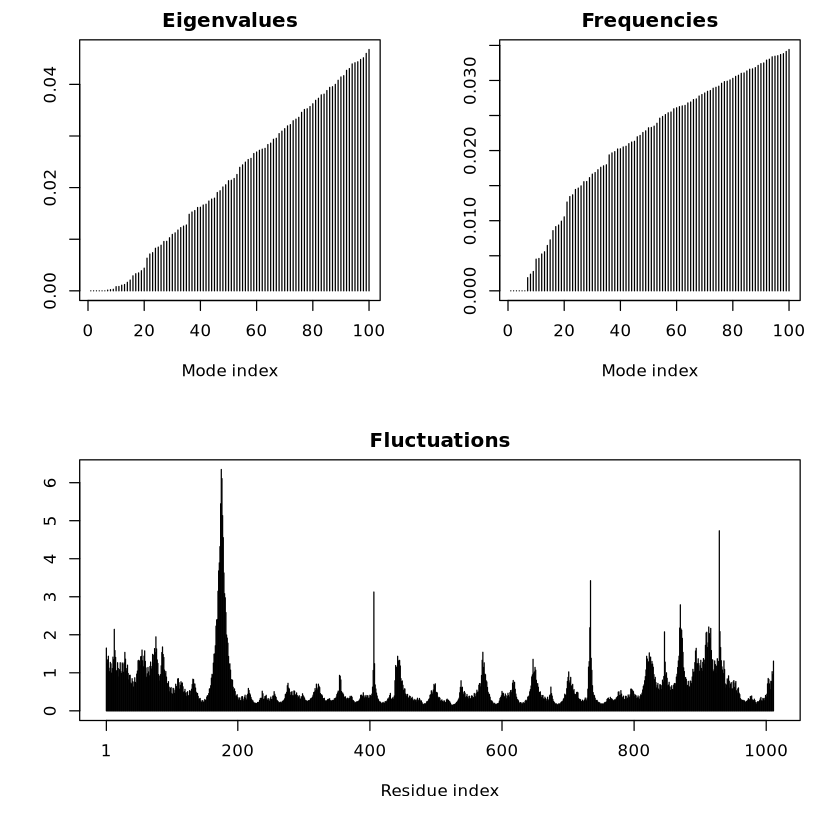
由圖可知,上方兩個圖分別顯示了特征值(Eigenvalues)和頻率(Frequencies)隨模式指數(Mode Index)的變化。特征值從左至右遞增,說明高模式對應的剛性較大,運動較難發生。頻率圖與特征值密切相關,也表現出遞增趨勢,高頻模式對應局部振動,低頻模式則與全局運動相關。下方的波動圖(Fluctuations)展示了蛋白質各殘基的振幅波動情況。某些殘基區域(如第185、400和730附近)顯示出較大的波動,意味著這些區域更具柔性,可能在蛋白質功能中起重要作用。
為了能夠清晰的展示蛋白質的柔性和其二級結構之間的關系,我們可以使用plot.bio3d()函數中的參數在波動圖(Fluctuations)上添加二級結構信息,
plot.bio3d(ctcf_mode$fluctuations, # NMA中每個殘基的波動數據 sse=ctcf, # 提供PDB對象以獲取蛋白質的二級結構信息sheet.col="orange", # 將β-折疊的顏色設置為橙色,用于在圖中區分二級結構helix.col="purple", # 將α-螺旋的顏色設置為紫色,用于區分二級結構typ="l", # 設置圖形類型為線條圖(line plot),即用線連接數據點lwd=3, # 將線條寬度設為3,使線條更加粗壯,圖形更清晰ylab="Fluctuations from NMA of CTCF-complex" # 設置y軸標簽為自定義內容
)
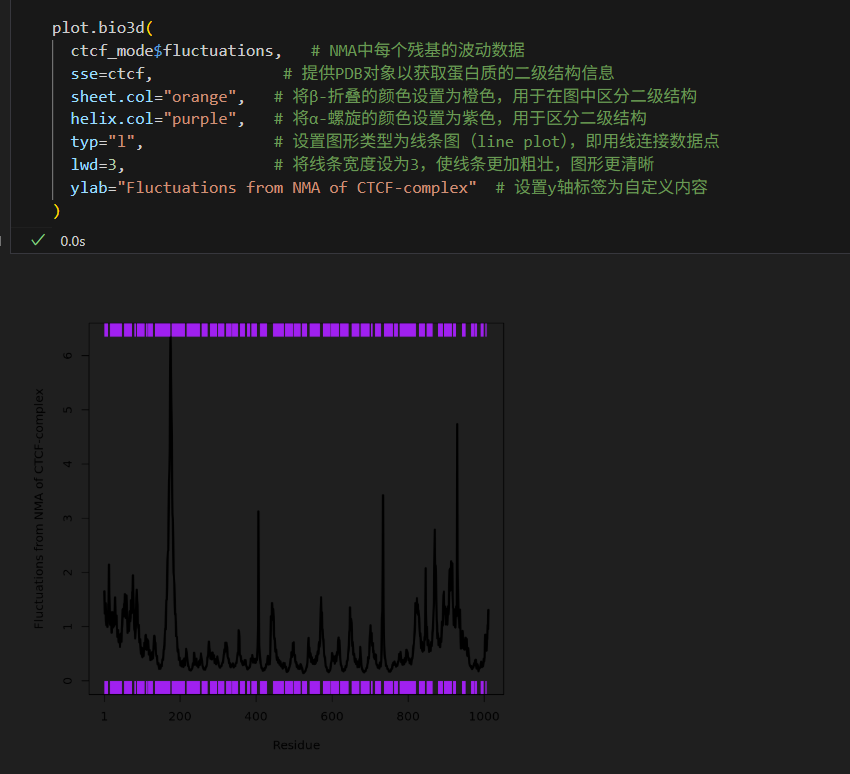

由圖可知,x軸表示殘基編號,y軸表示波動幅度。圖中的黑色曲線顯示了各個殘基的波動情況,波峰代表特定殘基有較大的波動,表明這些區域在蛋白質運動中柔性更高。紫色的橫條代表α螺旋區域,橙色橫條代表β折疊區域(當然圖中沒有,code中示意),這些區域的波動通常較小,表明它們結構較為穩定,柔性相對較低。圖像分析有助于研究蛋白質的動態特性,分析哪些區域更具柔性或穩定性。
2,2DFD分析示例
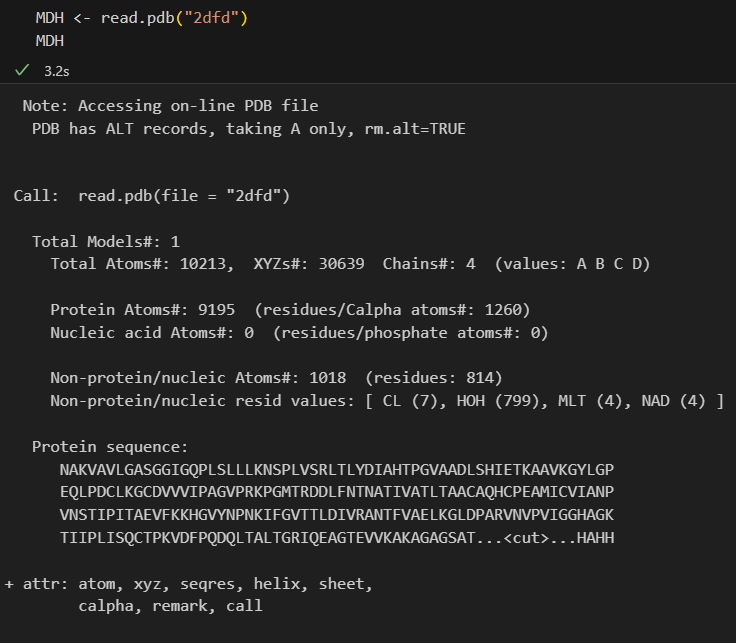
以蘋果酸脫氫酶為例,蘋果酸脫氫酶是一種在檸檬酸循環中催化草乙酸鹽和蘋果酸鹽轉化的同型二聚體酶,結構序列號為2DFD。

找出該結構中存在的鏈、原子和殘基的總數。計算結合位點并繪制酶中殘基的B因子:
這里解釋一下什么是B-factor:
蛋白質結構的b因子也稱為溫度結構,它表示蛋白質由于C α原子在其平均位置附近波動而產生的靈活性。由于這種靈活性,蛋白質分子的多肽骨架和側鏈會不斷運動。高b因子表明蛋白質分子中的殘基位置比平均值具有更高的柔韌性,而低b因子則反映了蛋白質分子中的剛性位置。隱藏在蛋白質分子核心的殘基可能具有較低的b值,因此與存在于蛋白質表面的殘基相比更具有剛性。
可以簡單將B-factor理解為柔韌性、剛性描述符。

plot.bio3d(MDH$atom$b[MDH$calpha], sse=MDH, typ="l", ylab="Bfactor")
帶有二級結構注釋


蘋果酸脫氫酶和乳酸脫氫酶是同源代謝酶。
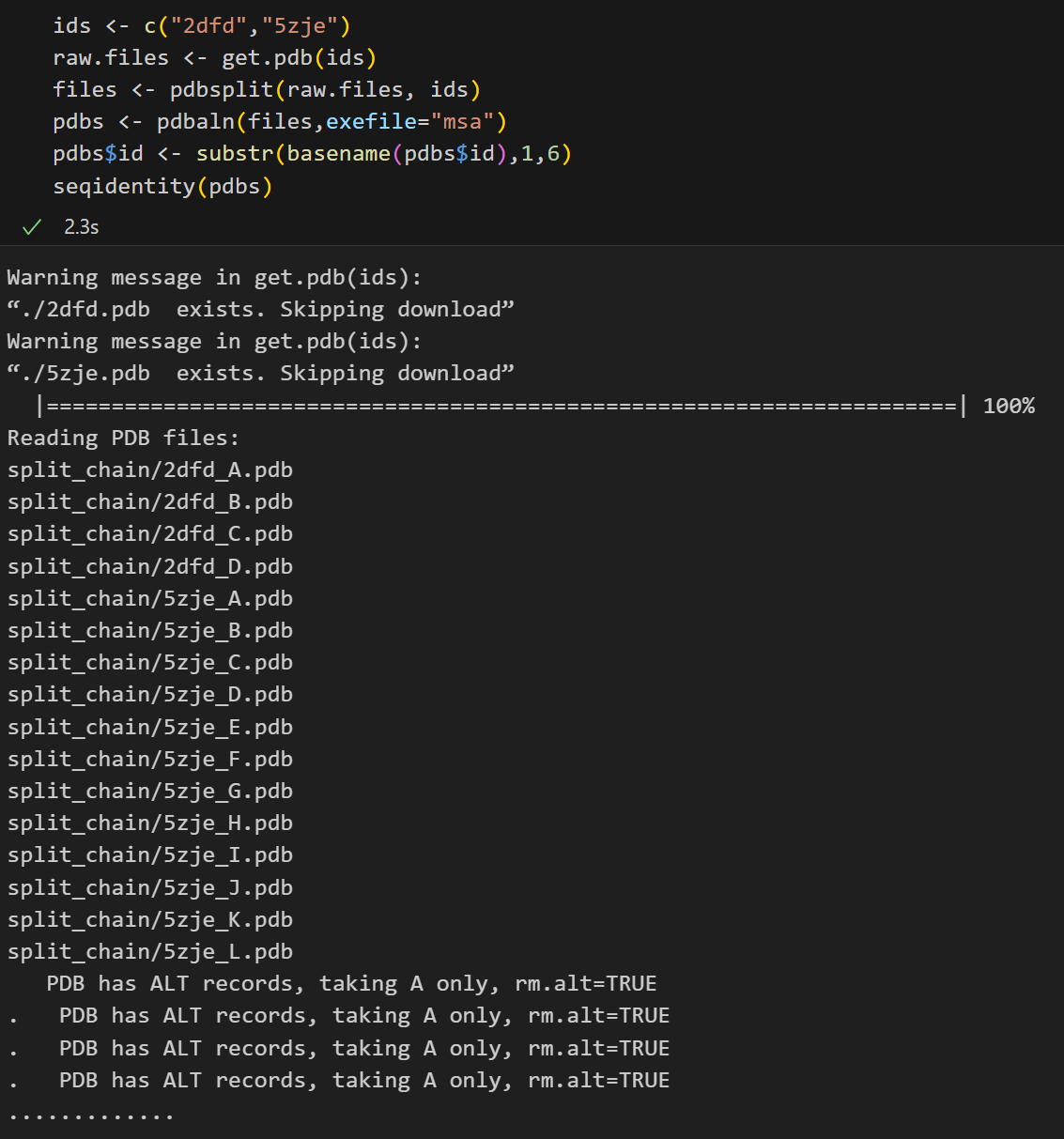
序列號:人蘋果酸脫氫酶(ID: 2DFD)和人乳酸脫氫酶(ID: 5ZJE)
(a)將存在于兩種脫氫酶結構中的鏈進行拆分和對齊,并計算蛋白質結構不同鏈之間的序列同一性和均方根偏差(RMSD)
(b)繪制RMSD直方圖并生成RMSD聚類的樹狀圖
ids <- c("2dfd","5zje")
raw.files <- get.pdb(ids)
files <- pdbsplit(raw.files, ids)
pdbs <- pdbaln(files,exefile="msa")
pdbs$id <- substr(basename(pdbs$id),1,6)
seqidentity(pdbs)
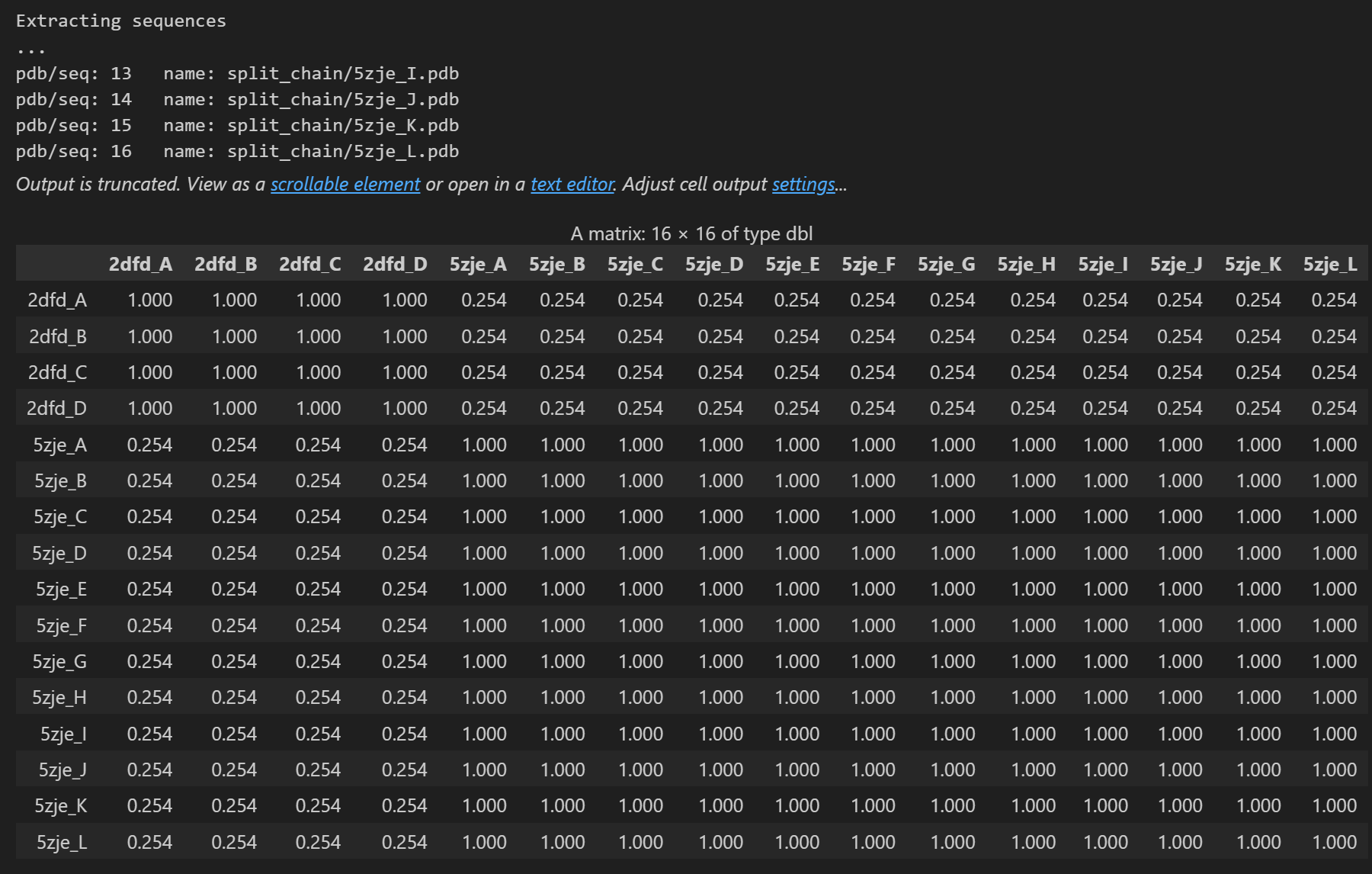
core <- core.find(pdbs)
core.inds <- print(core, vol=1.0)
xyz <- pdbfit( pdbs, core.inds )
rd <- rmsd(xyz)
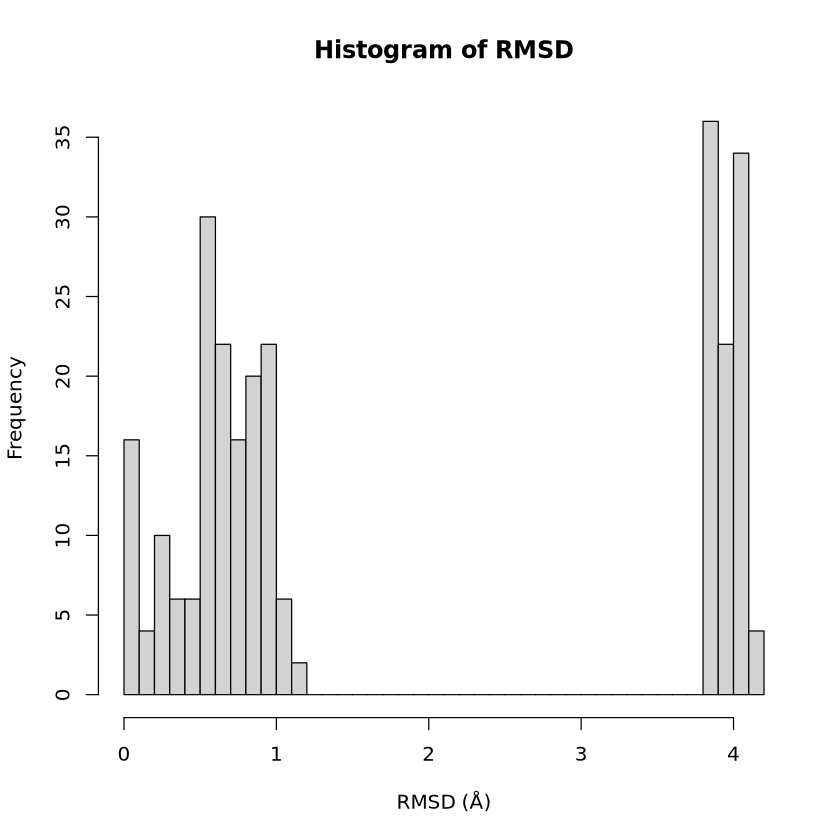
hist(rd, breaks=40, xlab="RMSD (A?)", main="Histogram of RMSD")
兩個酶不同鏈間RMSD的直方圖:
hc <- hclust(as.dist(rd))
hclustplot(hc, k=3, ylab="RMSD (A?)", main="RMSD Cluster Dendrogram")
基于蘋果酸脫氫酶和乳酸脫氫酶不同鏈間RMSD的RMSD聚類圖:



![文生3D實戰:用[靈龍AI API]玩轉AI 3D模型 – 第7篇](http://pic.xiahunao.cn/文生3D實戰:用[靈龍AI API]玩轉AI 3D模型 – 第7篇)




)









算法(附matlab代碼))

