一、寫在前面
基因捕獲效率、分辨率一直是空間轉錄組細胞類型識別的攔路虎,許多算法能夠整合單細胞(single-cell, sc)或單細胞核(single-nuclear, sn)數據與空間轉錄組數據,從而幫助空轉數據的細胞類型注釋。此前我們介紹過近年新出爐的Stereo-seq平臺,這一新興技術相關的分析思路及工具支持仍有所欠缺,因此本文的作者評估了9種單細胞數據與stereo-seq空轉數據進行mapping的算法,以幫助大家提示stereo-seq數據處理的準確性與效率。原文引用信息:
Tao Q, Xu Y, He Y, Luo T, Li X, Han L. Benchmarking mapping algorithms for cell-type annotating in mouse brain by integrating single-nucleus RNA-seq and Stereo-seq data. Brief Bioinform. 2024 May 23;25(4):bbae250.?
如果想系統性的學習空間轉錄組數據分析也可以看這里:
空間轉錄組學習手冊合輯
Stereopy空間轉錄組學習手冊
Squidpy空間轉錄組學習手冊
Scanpy空間轉錄組學習手冊
Seurat空間轉錄組學習手冊
一文搞定空間轉錄組與單細胞測序的整合分析
CellChat空轉細胞通訊合輯
SeekSpace| 會單細胞就會空間轉錄組
二、背景介紹
哺乳動物的腦組織具有極高的異質性以及復雜的細胞類型(神經元、免疫細胞、血管細胞等等)組成,較為傳統的技術例如免疫熒光或原位雜交技術無法同一時間內注釋出所有細胞類型,因此,腦組織常作為空間轉錄組想要占領的測試高地:長腦子了!時空轉錄組揭示大腦再生機制。stereo-seq憑借分辨率高(0.22μm)、視場大(芯片大小可定制)、無需預先設置探針等特點被廣泛應用于生命科學研究的各個領域之中。但實戰過程中,"cellbin"的劃分細胞策略容易遇到體積小的細胞捕獲到的分子數量不足、"square bin"劃分細胞不能滿足單細胞分辨率的尷尬。因此實際分析過程中stereo-seq與其它空間轉錄組數據一樣需要依賴單細胞水平的數據的mapping幫助完成注釋過程。目前能夠完成這一過程的軟件眾多:DestVI可以通過基因的"罰分"來完成參考單細胞數據與空轉數據的神經網絡構建;RCTD利用最大相似度預測各spot的細胞占比(能夠減少平臺不同帶來的影響);SpatialDWLS本質上利用阻尼加權最小二乘回歸,通過整合富集分析以及Giotto提供的差異分析來完成mapping過程;大家熟悉的神包Seurat能夠通過最近臨近法整合多來源的細胞;Tangram能夠梯度優化的計算連哥哥數據集間的KL離散度與余弦相似性;SpatialID利用轉化學習訓練深度神經網絡模型生成各細胞在空轉中分布的可能性;SpatialID利用每個數據集的高變基因構建系統模型,輸出全局最優的轉化矩陣可能性;Spann也能夠優化轉化模型,使得臨近的的樣本具有相同的細胞類型。此前也有過空轉與單細胞數據整合的工具benchmarking(空間轉錄組與單細胞轉錄組整合分析工具大比拼),但新出現的stereo-seq在之前并沒有被納入考察目標之中,并且這些工具的benckmarking大都依賴高質量的”groudtruth“,而stereo-seq能夠使用不同大小的bin size來互相做校驗,顯然對于這些工具來說也是一個良好的測試數據集。
三、主要結果
如Figure1 A的流程圖所示,作者通過已注釋的snRNA-seq數據生成了一個擬空間數據作為background,然后收集了八個包含對應腦區位置(例如海馬體、小腦、嗅球、皮質等)的snRNA-seq數據與stereo-seq數據。作者主要從square bin 50(大約25μmX25μm)與cell bin(利用ssDNA劃分生成的細胞單位)的各算法效果,可以明顯看出,Bin50的spot數量要明顯小于Cellbin,但nFeature和nCount方面前者更高,換句話說,在這些數據中Bin50的尺寸要大于Cellbin(Biomamba目前處理過的數據也基本都是這個情況)。
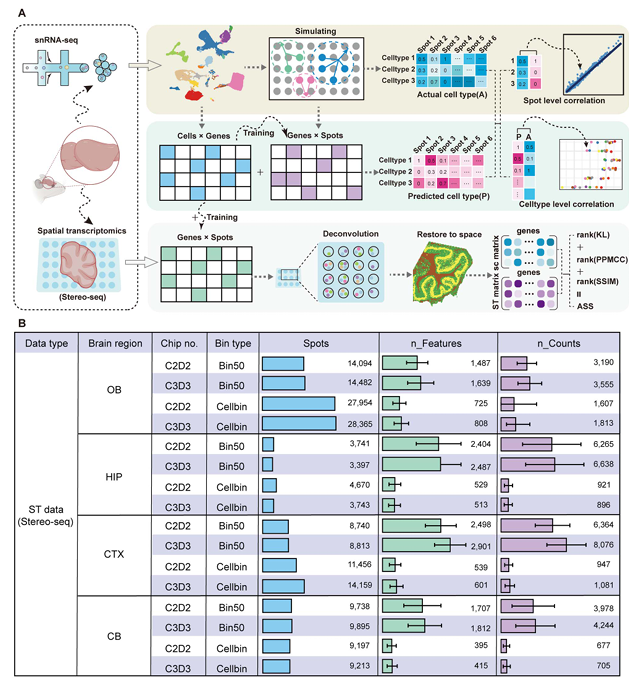
Figure 1
在完成了四個腦區數據的注釋(Figure 2A-B)之后,使用各mapping算法工具處理擬空間數據與stereo-seq空轉數據,并對每個spot中細胞比例進行皮爾森相關系數計算(Figure 2C),肉眼可見Cell2location(基于Python)與RCTD(基于R)的相關系數又高又穩(這兩個包的教程可見:一文搞定空間轉錄組與單細胞測序的整合分析)。在合并相同細胞類型的表達矩陣后,作者對stereo-seq細胞表達矩陣與snRNA-seq的細胞表達矩陣進行相關性系數分析(Figure 2D),可以看出RCTD這次遙遙領先,其余軟件除了Tangram外表現都很差。
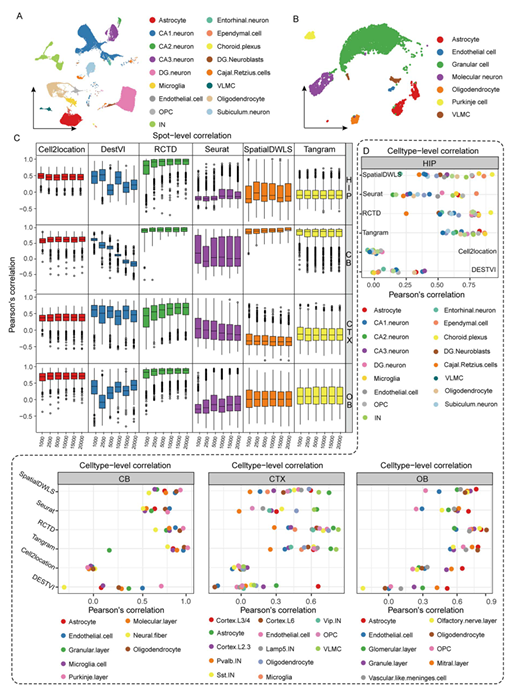
Figure 2
接下來作者開始了實戰部分,成年腦矢狀面腦組織HIP的snRNA-seq與stereo-seq的運行結果如圖3A所示,通過作者的生物學知識基礎,可以發現RCTD與SpatialDWLS對主要的細胞類型如CA1、CA2、CA3分布的預測較為精準,而其它軟件存在邊界不清晰、注釋不準確的現象。作者引入了ASS來評估細胞距離及相關性,結果顯示RCTD(在多個數據集及分bin方法中均表現上乘)與SpatialDWLS表現最優,而Spatial-ID與DestVI墊底(Figure 3B-C)。在注釋結果的細胞類型中可見,RCTD與SpatialDWLS能夠預測到占比非常小的細胞類型,而DestVI這種僅能夠預測到占比比較大的細胞類型。
ASS公式如下,感興趣的同學可以看一下原文了解詳細參數含義:
![]()
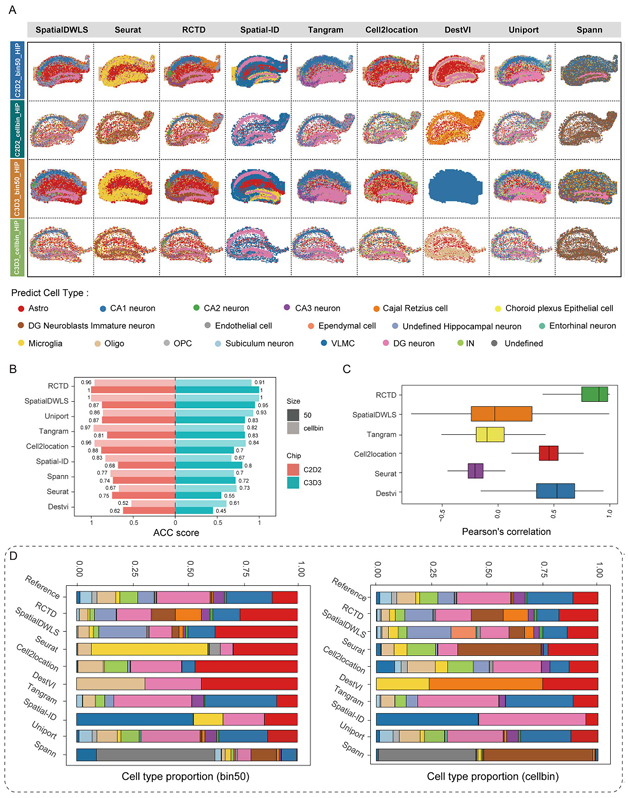
Figure3
在小腦數據的layer的識別上來說,RCTD與SpatialDWLS無論是在cell bin還是在bin50均能夠精準的分配出符合其空間位置的細胞類型(Figure 4A),而其它工具都或多或少的丟失了一些layer,例如Cell2location沒有識別出Purkinje cell layer。

Figure 4
四、最后聊聊
這篇文章想提供給大家的信息很簡單,如果你想用R語言處理stereo-seq,你就用RCTD,如果你想用python處理stereo-seq,你就用SpatialDWLS。值得一提的是,以上的結果都是作者使用腦部數據測試得到的結果,其它的組織器官可能會遇到不同的情況。并且,作為一個benchmarking的文章,作者并沒有給出計算效率的評估,要知道,不同算法/工具對于相同輸入數據的計算時間可能會相差數千倍。最重要的是作者提出了Accuracy scoring system(ASS)這一概念來評估各工具對空間轉錄組mapping的效果。本篇文章的代碼鏈接如下,大家可以自行學習體會一番:https://github.com/qyTao185/Benchmarking-Mapping-Algorithms.git
如果你的計算機不足以支持該工具的計算,可按需選用適合自己的計算資源:
共享(經濟實惠):有root權限的共享服務器
獨享(省電省心):生信分析不求人





)

:獨立部署是否拋棄了架構優勢?)
)










