生物信息學數據分析流程的搭建是一項繁重而復雜的工作。隨著行業的發展,各種生信流程框架層出不窮,比如有:
Nextflow
Snakemake
CWL
WDL
各種標準,各種規則,令人眼花繚亂。選擇太多,往往令人無所適從。特別是新進入行業的人,不知道從何著手,一個一個學過去,對時間精力的耗費是巨大的,也是不必要的。不僅如此,既有的流程框架為了追求大而全,往往顯得很笨重,這無疑增加了開發、維護和使用的難度。
針對現有流程框架的不足,我們開發了zflow,旨在為組學數據分析提供一個通用而又簡單易用的輕量級框架。
https://github.com/jianzuoyi/zflow功能簡介
支持單樣本流程搭建:如轉錄組標準分析、WGS、WES重測序等。
支持配對樣本流程搭建:如腫瘤NGS變異檢測等。
支持加測:即支持一個樣本多個文庫,一個文庫多條Lane的實驗設計。
設計理念
我們相信 Linux 是生信流程設計的通用語言,堅持直接用Linux shell腳本編寫生信流程。
整體框架
流程主要由兩部分組成:
模板文件:我們將流程所需的所有軟件/腳本調用,計算資源(Cpu, 內存)以及任務之間的依賴關系,在一個 XML 模板文件中定義。
解析器:zflow,負責將XML模板文件解析為Shell腳本。
投遞器:針對不同的集群環境,編寫相應的任務投遞插件。
zflow程序只有一個,即所有流程通用的。而每一條流程,會編寫一個XML模板文件。任務投遞,則是根據集群類型的不同,交給不同的投遞插件。
軟件依賴
Python 3.9.18
pandas 2.0.3
networkx 3.2.1
安裝好Python和依賴的軟件包,并將Python所在的路徑加入環境變量中,如:
export?PATH=/path/to/python3.9.18/bin:$PATH快速開始
git?clone?git@github.com:jianzuoyi/zflow.gitcd?zflow./zflow?-husage:?zflow?[-h]?--xml?XML?[--env?ENV]?[--bed?BED]?[--par?PAR]?--config?CONFIG?--outdir?OUTDIR?[--overwrite]?[--verbose]A?pipeline?framework?for?NGS?data?analysis.optional?arguments:-h,?--help???????show?this?help?message?and?exit--xml?XML????????XML?config?file?for?different?project?Type--env?ENV????????The?path?of?Environment.sh?[$PIPEDIR/config/Environment.sh]--bed?BED????????The?path?of?BED?file?[Default?is?$Target?in?Environment.sh]--par?PAR????????Parameters?file?[Para=Value?per?line]--config?CONFIG??Config?file?of?Data?path?and?Custom?Infomation--outdir?OUTDIR??Output?directory--overwrite??????Overwrite?output?results--verbose????????Show?detail?message使用說明
1. 生成任務
準備config文件:config.tsv
Project?Patient?Sample?Type?Data
mRNA?hg002_gm24385?hg002_gm24385?.?/ifs/public/test-data/giab/hg002_gm24385.mrna.R[12].fastq.gz
mRNA?hg002_gm26105?hg002_gm26105?.?/ifs/public/test-data/giab/hg002_gm26105.mrna.R[12].fastq.gz
mRNA?hg002_gm27730?hg002_gm27730?.?/ifs/public/test-data/giab/hg002_gm27730.mrna.R[12].fastq.gz
mRNA?hg004_gm24143?hg004_gm24143?.?/ifs/public/test-data/giab/hg004_gm24143.mrna.R[12].fastq.gz
mRNA?hg005_gm24631?hg005_gm24631?.?/ifs/public/test-data/giab/hg005_gm24631.mrna.R[12].fastq.gzProject: 項目名稱,可以任意設置
Patient: 病人/對象ID,可以重復,因為一個病人/對象可能有多個樣本
Sample: 樣本ID,一個病人/對象可以有多個樣本,但樣本ID必須唯一
Type:樣本類型,用于區分配對樣本,如果T代表腫瘤樣本,N代表正常對照樣本
Data: 數據路徑。支持雙端測序數據的分析。
準備運行腳本:run.sh
#!/bin/bash
set?-euo?pipefailconfig=./config.tsv
outdir=./result
template=./RNAseq_Standard_Analysis_PE.xml./zflow?\--xml?$template?\--config?$config?\--outdir?$outdir?\--verbose--xml ?流程模板文件
--config ?樣本信息配置文件
--outdir ?分析結果輸出目錄
--verbose ?解析XML過程中輸出詳細提示信息,否則采用安靜模式
運行:
./run.sh最后輸出:
...
Reading?tasks
Checking?task?dependencies
Sorting?tasks
Sorting?tasks?doneLocal?tasks:?/nas/develop/zflow/result/Tasks/mRNA/bin/mRNA_tasks.sh最終輸出的文件 mRNA_tasks.sh 即是總的Shell腳本文件,該文件中每一行是一個任務腳本。可以看到,每個任務腳本的文件名是以01,02這樣的數字編號開頭的,這些編號即表示該任務在整個流程中的順序。
less?mRNA_tasks.sh/nas/develop/zflow/result/Tasks/mRNA/bin/01_Begin_mRNA.sh/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg002_gm24385_hg002_gm24385.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg002_gm26105_hg002_gm26105.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg002_gm27730_hg002_gm27730.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg004_gm24143_hg004_gm24143.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg005_gm24631_hg005_gm24631.sh/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg002_gm24385.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg002_gm26105.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg002_gm27730.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg004_gm24143.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg005_gm24631.sh/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg002_gm24385_hg002_gm24385.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg002_gm26105_hg002_gm26105.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg002_gm27730_hg002_gm27730.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg004_gm24143_hg004_gm24143.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg005_gm24631_hg005_gm24631.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg002_gm24385.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg002_gm26105.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg002_gm27730.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg004_gm24143.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg005_gm24631.sh...2. 投遞任務
理論上來說,逐行執行 mRNA_tasks.sh 中的腳本,就可以得到分析結果,只是不能充分利用集群的計算資源。
你可以將這些任務手動投遞到集群上,編號相同的任務可以同時投遞。這是一種解決方案,但顯然不是最優的。更好的方案是寫一個插件與zflow銜接,將zflow生成的任務自動投遞到集群上。
這得益于zflow兩點獨有的設計:其在生成腳本時,同時保留了任務的依賴關系,以及每一個任務所需要的Cpu和內存資源。
mRNA_tasks.sh文件的后面,還跟著一些以#order開頭的行,這些行就表示任務之間的依賴關系。后續插件可以根據這些信息將所有任務添加到一個DAG圖當中,從而自動高效地向集群投遞任務。
#order?01_Begin_mRNA?before?02_Cat_Fastq_hg002_gm24385_hg002_gm24385
#order?01_Begin_mRNA?before?02_Cat_Fastq_hg002_gm26105_hg002_gm26105
#order?01_Begin_mRNA?before?02_Cat_Fastq_hg002_gm27730_hg002_gm27730
#order?01_Begin_mRNA?before?02_Cat_Fastq_hg004_gm24143_hg004_gm24143
#order?01_Begin_mRNA?before?02_Cat_Fastq_hg005_gm24631_hg005_gm24631#order?02_Cat_Fastq_hg002_gm24385_hg002_gm24385?before?03_Cat_Fastq_hg002_gm24385
#order?02_Cat_Fastq_hg002_gm26105_hg002_gm26105?before?03_Cat_Fastq_hg002_gm26105
#order?02_Cat_Fastq_hg002_gm27730_hg002_gm27730?before?03_Cat_Fastq_hg002_gm27730
#order?02_Cat_Fastq_hg004_gm24143_hg004_gm24143?before?03_Cat_Fastq_hg004_gm24143
#order?02_Cat_Fastq_hg005_gm24631_hg005_gm24631?before?03_Cat_Fastq_hg005_gm24631#order?03_Cat_Fastq_hg002_gm24385?before?04_FastQC_hg002_gm24385_hg002_gm24385
#order?03_Cat_Fastq_hg002_gm26105?before?04_FastQC_hg002_gm26105_hg002_gm26105
#order?03_Cat_Fastq_hg002_gm27730?before?04_FastQC_hg002_gm27730_hg002_gm27730
#order?03_Cat_Fastq_hg004_gm24143?before?04_FastQC_hg004_gm24143_hg004_gm24143
#order?03_Cat_Fastq_hg005_gm24631?before?04_FastQC_hg005_gm24631_hg005_gm24631
#order?03_Cat_Fastq_hg002_gm24385?before?04_Fastp_hg002_gm24385
#order?03_Cat_Fastq_hg002_gm26105?before?04_Fastp_hg002_gm26105
#order?03_Cat_Fastq_hg002_gm27730?before?04_Fastp_hg002_gm27730
#order?03_Cat_Fastq_hg004_gm24143?before?04_Fastp_hg004_gm24143
#order?03_Cat_Fastq_hg005_gm24631?before?04_Fastp_hg005_gm24631...你可能要問,那每一步的計算資源(Cpu,內存)需求在哪里呢?可以打開任意一個腳本,如:
less?/nas/develop/zflow/result/Tasks/mRNA/bin/05_Align_Hisat2_hg002_gm24385.sh#!/bin/bash
set?-euxo?pipefail
source?/pub/pipeline/RNAseq/config/Environment.sh
Cpu=8
Mem=32cd?/nas/develop/zflow/result/mRNA/hg002_gm24385/hg002_gm24385/02.Aln
hisat2?\-x?$REFERENCE_GENOME?\-1?../01.QC/hg002_gm24385_clean_R1.fq.gz?\-2?../01.QC/hg002_gm24385_clean_R2.fq.gz?\--summary-file?hg002_gm24385_summary.txt?\--new-summary?\-p?8?\--dta?-t?|\
samtools?view?-Sb?>?hg002_gm24385.bamln?-s?`pwd`/hg002_gm24385_summary.txt?/nas/develop/zflow/result/mRNA/multiqc/multiqc_WDir/hisat2/可以看到,腳本的前面有:Cpu=8, Mem=32,即是指示該步驟需要8條線程,32G內存。負責投遞任務的插件可以讀取這兩個信息用于任務投遞。順便提一下,任務腳本開始有一行:
source?/pub/pipeline/RNAseq/config/Environment.sh即是導入環境變量,任務中所需的軟件的路徑應該在這個Environment.sh文件中定義好,如本任務所需要的 hisat2, samtools應該在Environment.sh文件中將其所在的目錄加入PATH。
最后來看一下腳本目錄的全家福:
01_Begin_mRNA.sh
02_Cat_Fastq_hg002_gm24385_hg002_gm24385.sh
02_Cat_Fastq_hg002_gm26105_hg002_gm26105.sh
02_Cat_Fastq_hg002_gm27730_hg002_gm27730.sh
02_Cat_Fastq_hg004_gm24143_hg004_gm24143.sh
02_Cat_Fastq_hg005_gm24631_hg005_gm24631.sh
03_Cat_Fastq_hg002_gm24385.sh
03_Cat_Fastq_hg002_gm26105.sh
03_Cat_Fastq_hg002_gm27730.sh
03_Cat_Fastq_hg004_gm24143.sh
03_Cat_Fastq_hg005_gm24631.sh
04_FastQC_hg002_gm24385_hg002_gm24385.sh
04_FastQC_hg002_gm26105_hg002_gm26105.sh
04_FastQC_hg002_gm27730_hg002_gm27730.sh
04_FastQC_hg004_gm24143_hg004_gm24143.sh
04_FastQC_hg005_gm24631_hg005_gm24631.sh
04_Fastp_hg002_gm24385.sh
04_Fastp_hg002_gm26105.sh
04_Fastp_hg002_gm27730.sh
04_Fastp_hg004_gm24143.sh
04_Fastp_hg005_gm24631.sh
05_Align_Hisat2_hg002_gm24385.sh
05_Align_Hisat2_hg002_gm26105.sh
05_Align_Hisat2_hg002_gm27730.sh
05_Align_Hisat2_hg004_gm24143.sh
05_Align_Hisat2_hg005_gm24631.sh
05_FastQC2_hg002_gm24385.sh
05_FastQC2_hg002_gm26105.sh
05_FastQC2_hg002_gm27730.sh
05_FastQC2_hg004_gm24143.sh
05_FastQC2_hg005_gm24631.sh
06_Align_Hisat2_Sort_hg002_gm24385.sh
06_Align_Hisat2_Sort_hg002_gm26105.sh
06_Align_Hisat2_Sort_hg002_gm27730.sh
06_Align_Hisat2_Sort_hg004_gm24143.sh
06_Align_Hisat2_Sort_hg005_gm24631.sh
07_Stringtie_hg002_gm24385.sh
07_Stringtie_hg002_gm26105.sh
07_Stringtie_hg002_gm27730.sh
07_Stringtie_hg004_gm24143.sh
07_Stringtie_hg005_gm24631.sh
08_Stringtie_preDE_mRNA.sh
09_MultiQC_mRNA.sh
10_End_mRNA.sh
mRNA_tasks.sh一個總的腳本文件mRNA_tasks.sh,記錄所有要執行的任務以及任務之間的依賴關系。
多個任務腳本文件,記錄著每一個任務要執行的命令以及所需要的計算資源。
模板簡介
流程的描述信息我們統一放到一個XML模板文件當中。該模板的結構為:
<Pipeline>
<Task><Name>Align_Hisat2_$Sample</Name><Rely>Fastp_$Sample</Rely><Cpu>8</Cpu><Mem>32</Mem><Workdir>$Outdir/$Project/$Patient/$Sample/02.Aln</Workdir><Command><![CDATA[hisat2?\-x?$REFERENCE_GENOME?\-1?../01.QC/$Sample_clean_R1.fq.gz?\-2?../01.QC/$Sample_clean_R2.fq.gz?\--summary-file?$Sample_summary.txt?\--new-summary?\-p?$Cpu?\--dta?-t?|\samtools?view?-Sb?>?$Sample.bamln?-s?`pwd`/$Sample_summary.txt?$Outdir/$Project/multiqc/multiqc_WDir/hisat2/]]></Command>
</Task><ENV>
<Environment>/path/to/RNAseq/config/Environment.sh</Environment>
</ENV>
</Pipeline>每一個配對的<Task>,</Task>標簽內是一個任務的描述。其中包含:
Name:任務名稱
Rely:任務依賴的步驟名稱
Cpu:所需Cpu資源
Mem:所需內存資源
Workdir:工作目錄
Command: 任務命令
其中以美元符號$或者@開頭的為變量($表示普通變量,@表示數組),在任務解析過程中將會被zflow依據輸入的參數信息替換為具體的值。詳情請參考流程示例文件:RNAseq_Standard_Analysis_PE.xml。
Environment.sh文件的作用前述已提及,用于放置所有環境變量。
流程設計
流程搭建非常容易,直接在XML中寫Shell代碼就可以了。但是zflow流程有一些保留的變量,或者說關鍵字,需要用戶事先掌握。請放心,關鍵字的數量并不多,并且都非常容易理解。
XML標簽中的關鍵字:
Name, Rely, Cpu, Mem, Workdir, Command,各關鍵字的作用前述也提及。
來源于命令行的關鍵字:
$Config:配置文件,即zflow的--config參數所指定的文件
$Target:BED格式文件,如WES或Panel測序中的探針捕獲區域文件
$Outdir:輸出目錄
來源于配置文件中的關鍵字:
總的來說,配置文件中的每一列,會成為一個變量傳遞給XML,另外一些數組可以通過計算變換獲得。
$Project:項目名稱
$Patient:病人/對象ID
:樣本,如果是腫瘤樣本,可以是Tumor表示腫瘤樣本ID, $Normal表示正常樣本ID
$Datadir:測序數據的原始位置
$Lib:文庫名稱
$Lane:Lane名稱
@Lib:數組,代表某一樣本的所有文庫
@Lane:數組,代表某一文庫的所有Lane
@Sample:數據,代表所有樣本ID
@Tumor:數組,代表所有腫瘤樣本ID
@Normal:數組,代表所有正常樣本ID
需要說明的是,配置文件中列出的5個列是強制要求的:Project Patient Sample ?Type ? ?Data,除此之外,你還可以在其中增加列,這些列名同樣會作為變量傳遞給XML。
更多變量想傳遞進XML,還可以通過 zflow 的 --par 參數實現:
--par?PAR????????Parameters?file?[Para=Value?per?line]在這個參數文件中,每一行定義一個變量,其格式為:
參數名稱=參數對應的值(可以是某個數值,或者文件的路徑等)可以通過配置文件config.tsv和專門的參數文件--par靈活地向XML文件傳遞變量,這有效地保證了zflow的靈活性。比如,你可以在config.tsv文件中增加一列,表示樣本分組。也可以將分組信息單獨放在一個文件中,再在--par文件中指明分組文件所有的路徑,這都是非常靈活的。
特點總結
綜上可以看出zflow設計生信流程的特點:
簡單易用。流程的所有設計都在XML文件中,zflow只負責解析XML,針對不同的流程,zflow無須做任何更改。
輕量級。zflow由單個Python腳本構成,僅有600多行代碼。
輕松解決任務之間的依賴關系。zflow能夠在生成任務腳本的過程中順便生成任務之間的依賴關系。開發人員所需要做的工作僅僅是在XML文件中指定一下某個任務需要依賴于哪個或哪些任務即可。更進一步,如果任務關系指定錯誤,zflow能夠給出錯誤提醒。
模塊化設計。zflow被設計成只負責生成Shell腳本,后續的集群任務投遞交給專用的插件完成。這極大地保證了流程框架的可擴展性。
很多生信流程,值得用zflow重寫一遍。
許可證
zflow 支持學術免費使用,歡迎 Fork、加星和下載。如需商用,請聯系【簡說基因】獲得授權。
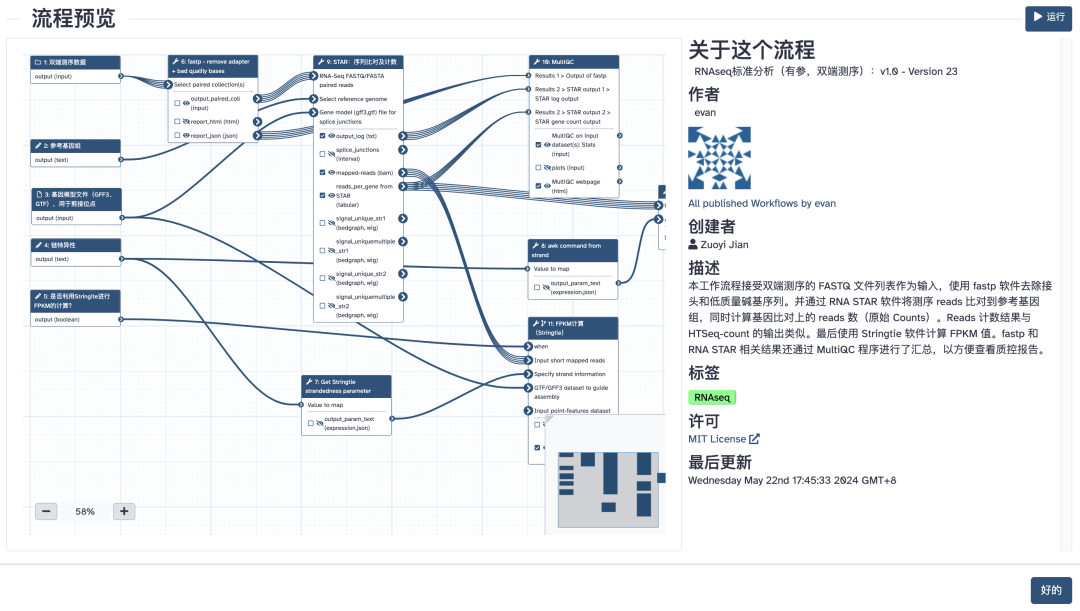
云上轉錄組分析流程(點擊圖片跳轉)
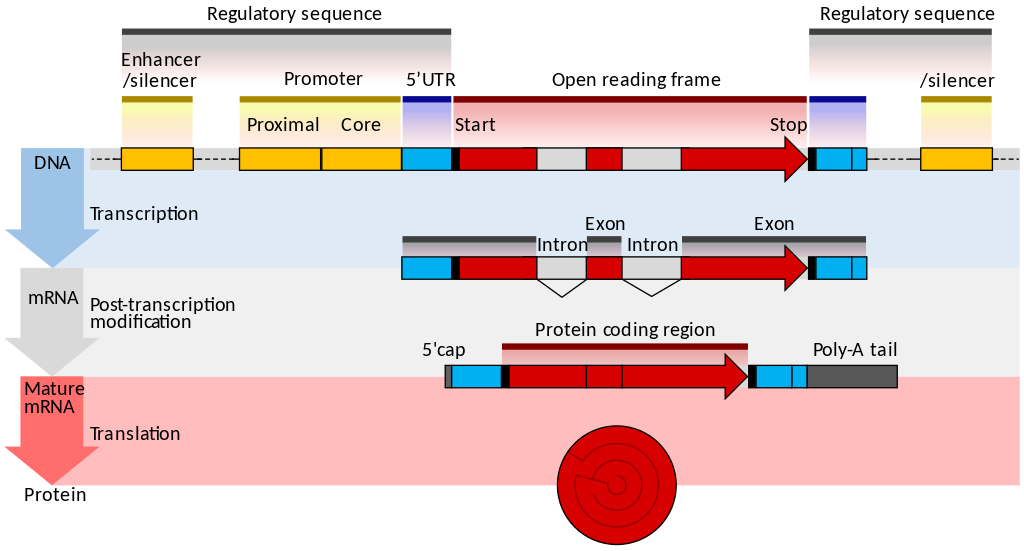
一文讀懂scRNA-seq數據分析(點擊圖片跳轉)
如何自學生物信息學:從菜鳥到專家(點擊圖片跳轉)

往期精彩:
生信人的自我修養:Linux 命令速查手冊
經典教程:全轉錄數據分析實戰
網上最全的 R 語言圖庫(建議收藏)| 簡說基因 Recommend
清華大學生物信息學課件資料分享
生物信息學軟件:兩種風格
新年第一課:從零開始入門Galaxy生信云平臺
生物信息學必備的R語言相關參考書 | 簡說基因 Recommand
從單細胞數據分析的最佳實踐看R與Python兩個陣營的博弈
涉嫌侵權,容我解(jiao)釋(bian)一下
生物信息學中的可重復性研究
關于簡說基因
生信平臺
Galaxy中國(UseGalaxy.cn)致力于打造中國人的云上生物信息基礎設施。大量在線工具免費使用。無需安裝,用完即走。活躍的用戶社區,隨時交流使用心得。
聯系方式
QQ交流群(免費):925694514
微信交流群(免費):加微信好友,注明“Galaxy交流群”
客服微信:usegalaxy







)







![[C語言]自定義類型詳解:結構體、聯合體、枚舉](http://pic.xiahunao.cn/[C語言]自定義類型詳解:結構體、聯合體、枚舉)
)


】)