
中國醫科大借iMeta影響因子躍升至33.2(中科院1區)東風,憑多組學聯合生信分析成果登刊
2025年6月18日,科睿唯安正式發布了2024年期刊的最新影響因子(發送關鍵詞 “2024JIF” 至 “生信學術縱覽”,即可下載完整版最新影響因子 Excel 表格!),這一消息迅速在學術界引發廣泛關注。影響因子作為衡量學術期刊影響力的重要指標之一,其每一次變動都如同在學術領域投入一顆石子,激起層層漣漪。眾多國際知名期刊的影響因子有了顯著變化,如腫瘤神刊 CA: A Cancer Journal for Clinicians 再度暴跌,四大醫學期刊繼續全線下跌;而 Science 則逆勢上漲。在這之中,值得一提的是,國產期刊 iMeta 表現十分亮眼,其影響因子從 2023 年的 23.8 暴漲 40%,一舉到達 33.2 的高分,成績斐然。這一變化不僅體現了 iMeta 期刊影響力的大幅提升,也彰顯了中國學術期刊在國際舞臺上日益重要的地位。在這樣充滿變化與活力的學術環境下,接下來將為大家介紹一篇發表于 iMeta 期刊的論文,讓我們一同深入探究其蘊含的學術價值 。
?https://doi.org/10.1002/imt2.70038

正式介紹
基本信息
-
論文標題:使用單細胞分析平臺對單細胞多組學數據進行綜合分析
-
發表期刊:iMeta,中科院微生物學大類分區1區Top,IF=33.2000
-
發表日期:2025年4月28日在線發表
研究背景
單細胞組學技術發展使研究能深入到單細胞層面,但現有分析平臺存在局限,如多針對單一模態數據,且使用復雜,需要大量編碼知識,難以滿足當前多組學數據分析需求,尤其對于流行的10× Genomics組學數據類型,缺乏全面且無需編碼的解決方案。
研究思路
開發這一基于網絡的平臺,整合多種先進算法、交互可視化功能和直觀界面,支持多種單細胞組學數據類型及空間轉錄組學分析。通過自動化關鍵分析步驟、提供豐富分析工具和可視化結果,降低分析門檻,使研究人員無需編程技能即可進行綜合分析。
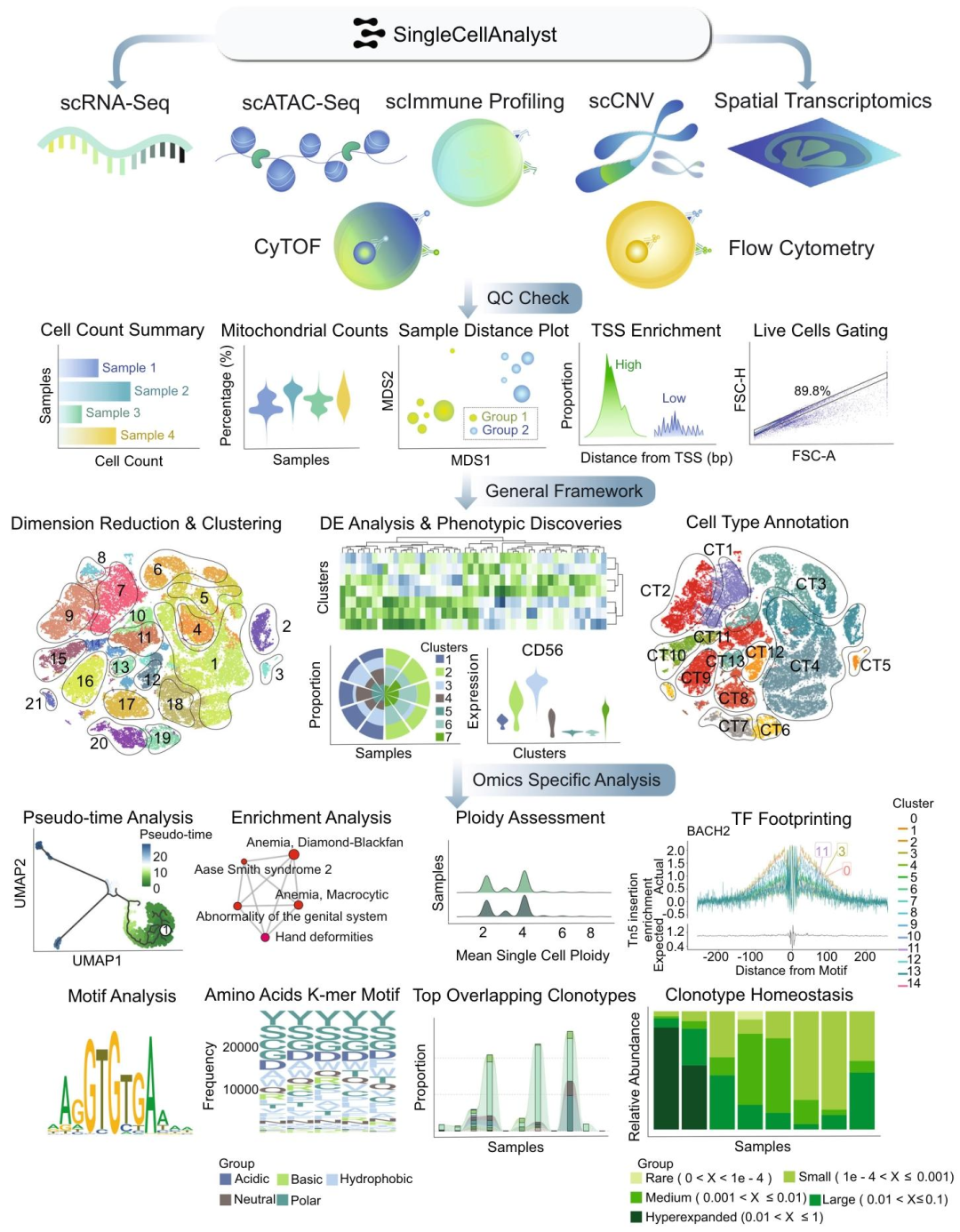
圖1:單細胞分析平臺網頁服務器上的分析框架概述

圖2:單細胞分析平臺數據庫概述
研究亮點
多組學覆蓋全面:支持六種單細胞組學類型和空間轉錄組學,涵蓋幾乎所有前沿單細胞組學技術,遠超其他同類平臺。操作簡便易用:無需編碼,界面直觀友好,簡化工作流程,降低學習成本,使不同技術背景的研究人員都能輕松使用。功能豐富強大:自動化質量控制、數據處理和表型分析等關鍵步驟,提供20多種交互式分析工具,可進行綜合分析和可視化,且分析速度快,能高效利用計算資源。數據連接廣泛:與單細胞圖譜數據庫緊密連接,包含多種組學數據,為比較分析提供豐富資源。
數據來源和生物信息方法
1、數據來源
包括多種單細胞組學數據,如單細胞RNA測序(scRNA - seq)的基因 - 細胞計數矩陣(.csv或.txt格式,或10X Genomics的.h5格式)、單細胞染色質可及性測序(scATAC - seq)的片段文件(.bed)等,還有空間轉錄組學的表達矩陣(.h5或.csv)及相關元數據等。
2、生物信息方法
使用R語言(v4.2.0)和shiny應用進行前后端集成,依賴seurat(v4.1.0)、Monocle3(v1.0.0)等多種R包。采用主成分分析(PCA)、t - 分布隨機鄰域嵌入(t - SNE)、均勻流形近似和投影(UMAP)等算法進行降維;k - 最近鄰(k - NN)、共享最近鄰(SNN)模塊化優化算法進行聚類;Wilcoxon秩和檢驗、邏輯回歸等進行差異表達分析;基因集富集分析(GSEA)等進行通路富集分析。
主要結果
1、scRNA-seq分析結果
完成質量控制、數據處理后,進行細胞類型預測、差異表達分析、通路和富集分析以及偽時間軌跡分析。多樣本時可進行綜合分析,發現表型差異。例如在研究腫瘤微環境異質性時,可通過聚類識別不同細胞群體,差異表達分析找出相關基因,富集分析揭示功能機制。小結:scRNA - seq分析功能全面,能深入研究基因表達動態和細胞狀態轉變。

圖3:scRNA-seq框架的分析工作流程
2、scATAC-seq分析結果
質量控制包括轉錄起始位點(TSS)富集等檢查,后續進行可及染色質區域識別、基序足跡分析等。多樣本時整合分析揭示染色質特征。如研究細胞分化過程中,可通過分析染色質可及性與基因表達關系,探索調控元件作用。小結:scATAC - seq分析有助于深入了解染色質調控機制。

圖4:scATAC-seq框架的分析工作流程
3、scImmune Profiling分析結果
進行克隆型豐度等質量控制評估,開展免疫庫重疊分析、基因使用分析等。多模態數據時整合scRNA-seq數據,研究免疫反應。如在癌癥免疫治療研究中,可分析克隆型擴張與細胞類型關系。小結:scImmune Profiling分析為免疫相關研究提供詳細信息。

圖5:scImmune Profiling框架的分析工作流程
4、scCNV分析結果
進行倍性評估等分析,識別細胞亞群的基因組異常。多樣本時進行系統發育分析和層次聚類,研究CNV模式。在癌癥研究中,可用于檢測腫瘤細胞的基因組變化。小結:scCNV分析對研究癌癥等疾病的基因組變異有重要意義。

圖6:單細胞拷貝數變異(scCNV)框架的分析工作流程
5、CyTOF分析結果
處理原始.fcs文件,進行降維和聚類分析,識別細胞表型,包括標記物表達分析、特征選擇等。如研究復雜疾病的免疫細胞異質性時,可通過分析細胞表型差異,了解疾病機制。小結:CyTOF分析有助于深入研究細胞表型特征。
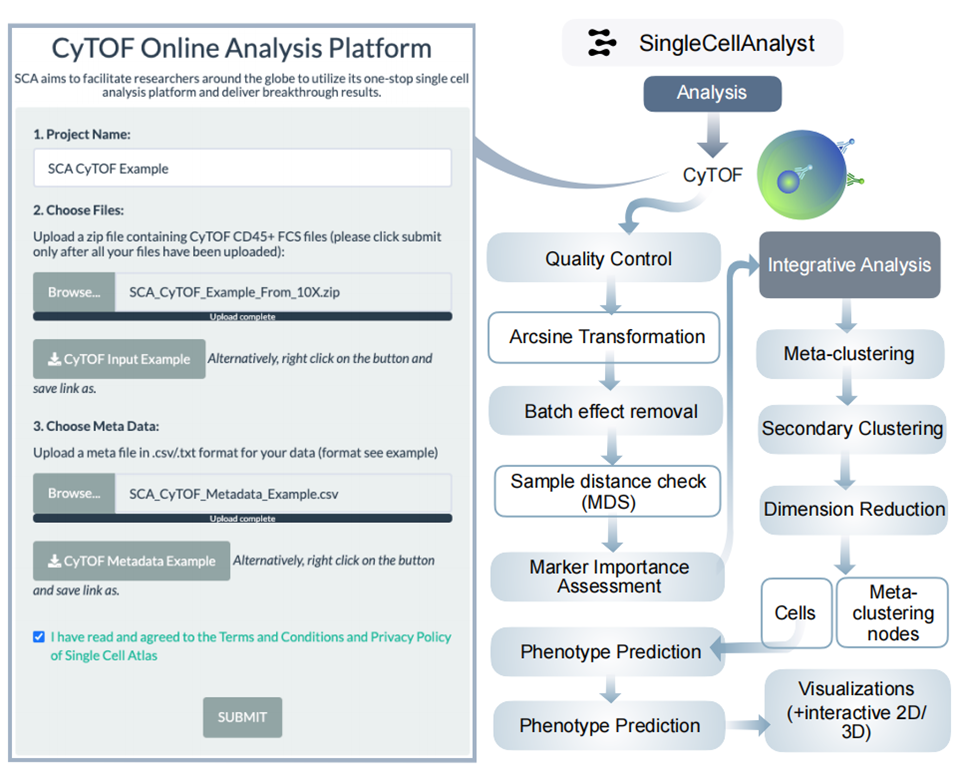
圖S4:CyTOF框架的分析工作流程
6、Flow cytometry分析結果
自動化門控策略去除雜質和雙峰,進行降維和聚類,評估標記物表達。多樣本時綜合分析群體差異,可用于免疫監測和生物標志物發現。小結:Flow cytometry分析能精準進行細胞表型分析。

圖S5:流式細胞術框架的分析工作流程
7、Spatial transcriptomics分析結果
進行質量控制和數據處理后,進行差異表達分析,將降維和聚類結果可視化在組織圖像上。多樣本時進行整合分析,結合scRNA - seq數據進行多模態整合。如研究組織發育時,可直觀展示基因表達的空間分布。小結:Spatial transcriptomics分析為研究組織空間結構和基因表達關系提供有力工具。
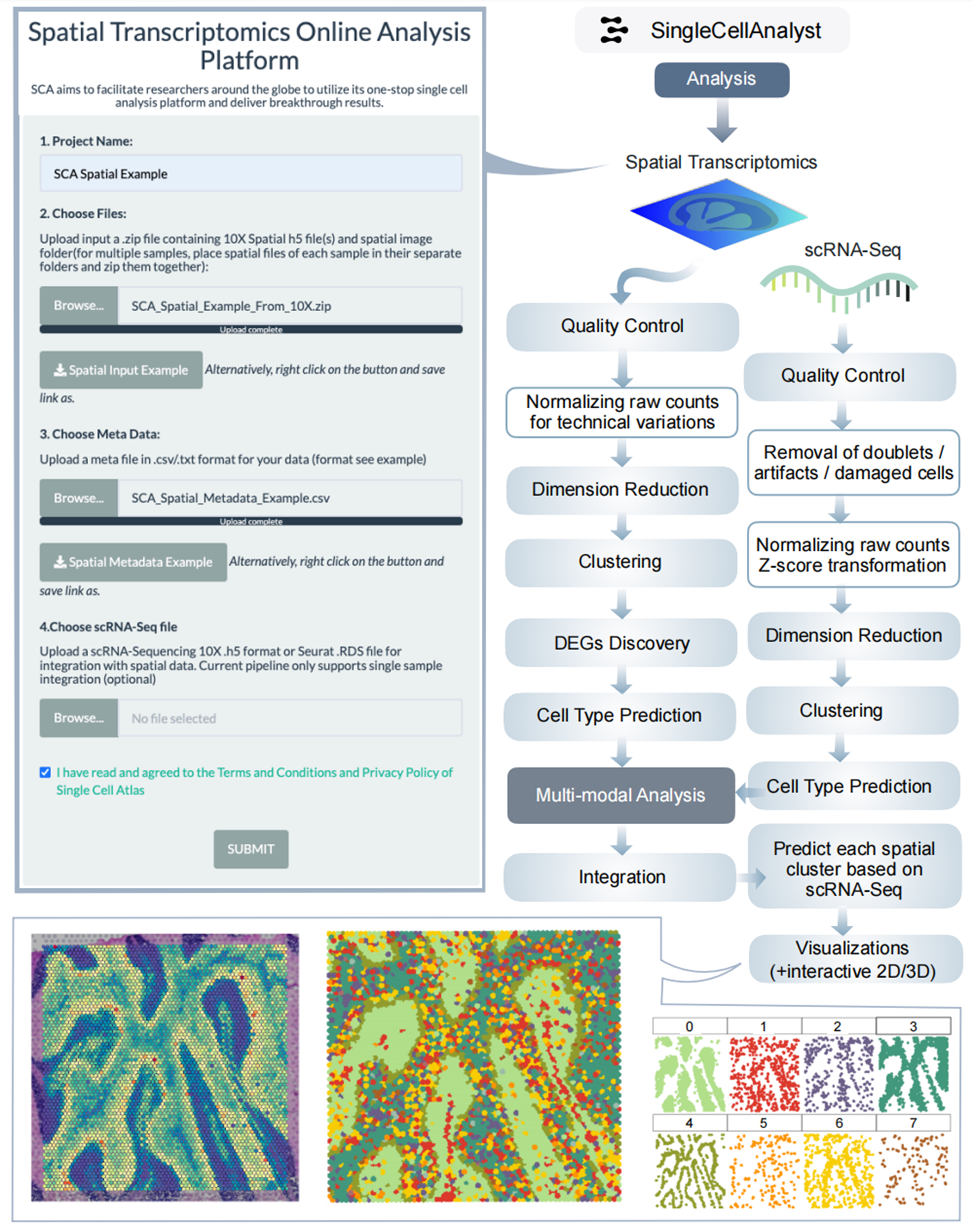
圖S3:空間轉錄組學框架的分析工作流程
研究結論
single-cell analyst支持多種單細胞組學和空間轉錄組學分析,簡化工作流程,自動化關鍵步驟,提供交互式可視化結果。降低了技術門檻,促進多組學分析,有助于理解生物現象,推動單細胞技術在生物和醫學研究中的應用,加速精準醫學發展。
研究的局限性和未來方向
目前主要針對人類數據分析,未來計劃支持更多物種;加強多模態單細胞數據集聯合分析能力,雖docker版本技術上可行,但當前版本為保證一致性暫未實現,后續更新將拓展;增加對更多組學數據類型(如代謝組學)的支持,強化數據整合和預測建模能力,提升計算可擴展性,根據用戶反饋持續改進。
感謝您的閱讀,歡迎關注“生信學術縱覽”。謝謝您的分享、點贊+在看!









)










