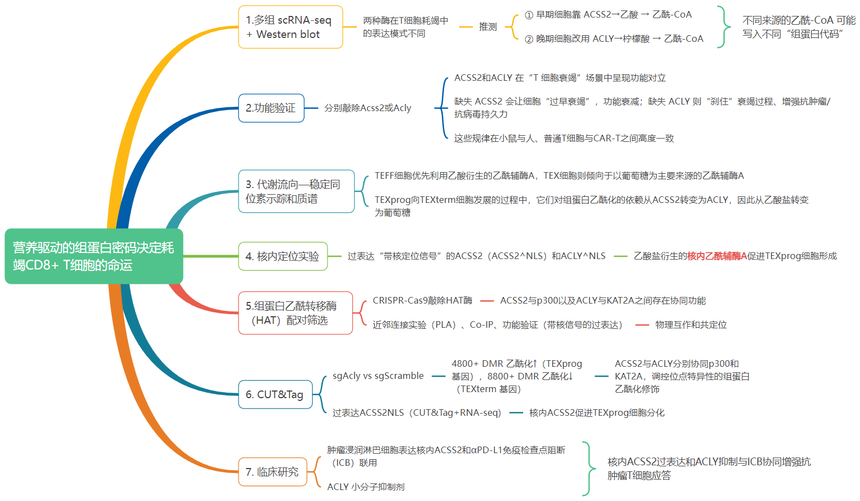
CD8?T細胞耗竭(T cell exhaustion)是腫瘤免疫治療的核心瓶頸,其表觀遺傳重塑機制(如組蛋白修飾)是當前國自然重點資助的前沿方向。耗竭T細胞(TEX)是指在慢性感染(如持續性病毒感染)或腫瘤微環境中長期暴露于抗原刺激后,功能逐漸失調的CD8?T細胞。研究顯示其在癌癥和慢性病毒感染中經歷代謝和表觀遺傳重塑,削弱了其保護能力。然而,營養代謝對控制TEX分化的表觀遺傳修飾的影響尚不清楚。今天我們帶來一篇Science文章《Nutrient-driven histone code determines exhausted CD8+ T cell fates》,該研究針對腫瘤免疫治療的核心瓶頸——CD8+T細胞耗竭(TEX)展開,首次揭示代謝重編程通過營養依賴的組蛋白修飾決定T細胞分化命運。

-
研究技術:RNA-seq、CUT&Tag、scRNA-seq等(愛基百客均可提供)。
-
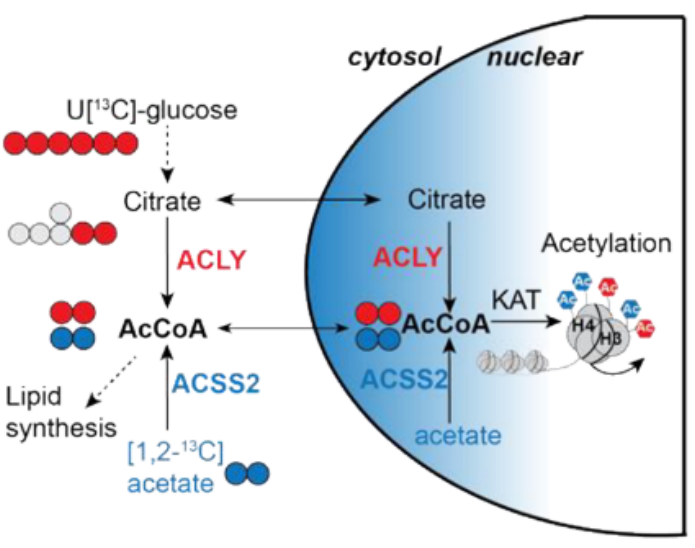
核心問題:本研究揭示了乙酰輔酶A合成酶ACSS2和ATP-檸檬酸裂解酶ACLY通過介導不同營養源(乙酸鹽vs.葡萄糖)驅動的組蛋白乙酰化,決定TEX細胞分化命運的關鍵機制。
? ?研究思路 ??
???研究結果???
1.?ACSS2與ACLY在TEFF(效應T細胞)和TEX細胞中的差異表達模式
為了理解選擇性營養物質利用及隨后的乙酰輔酶A(acetyl-CoA)生成如何調節CD8+T細胞分化,研究分析了從腫瘤或急性(Armstrong)與慢性(克隆13)淋巴細胞性脈絡叢腦膜炎病毒(LCMV)感染中分離出的CD8+T細胞中乙酰輔酶A合成酶Acss2和ATP-檸檬酸裂解酶Acly的轉錄譜(公共單細胞轉錄組數據,圖1A和1B)。另外,研究還從蛋白水平關注了ACSS2與ACLY的表達情況(圖1C和1D)。結果表明,隨著TEX細胞在腫瘤和慢性感染中發展,它們下調ACSS2表達同時維持ACLY表達。這里研究團隊發現了這兩種酶在T細胞耗竭中的表達模式不同。
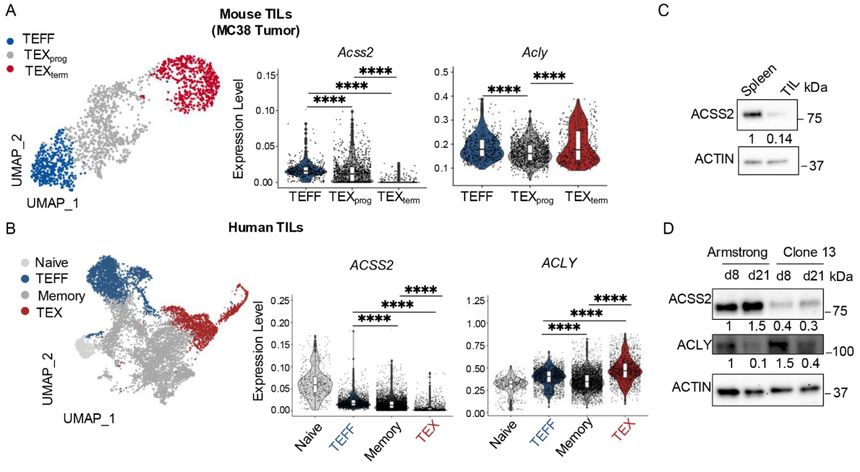
圖1:ACSS2表達下調,而ACLY表達在TEX細胞中持續存在。
2.?ACSS2和ACLY在TEFF和TEX細胞分化及抗腫瘤和抗病毒免疫中的功能拮抗
使用基因敲除方法研究了ACSS2和ACLY在腫瘤發生和慢性LCMV感染期間TEX細胞形成中的作用。在導致T細胞耗竭的情況下,Acss2的缺失削弱了CD8+T細胞活性,而Acly缺失則增強了它們的功能。
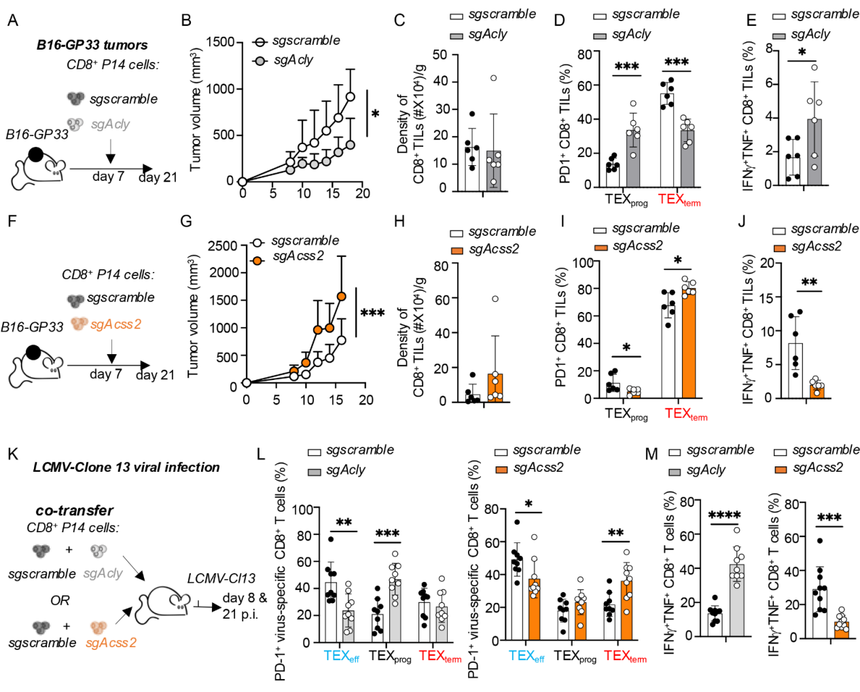
圖2:ACSS2促進TEXprog細胞形成及抗腫瘤/抗病毒反應,而ACLY則起抑制作用。
3.?ACSS2與ACLY決定了TEFF和TEX細胞對底物的選擇性利用
ACSS2和ACLY的差異表達模式及其對TEX細胞形成的影響讓研究團隊提出假設,即乙酸鹽和葡萄糖衍生檸檬酸鹽合成乙酰輔酶A的過程可能在功能性效應T細胞和功能障礙性耗竭T細胞之間存在差異。于是,研究團隊使用穩定同位素示蹤實驗來追蹤乙酰輔酶A的來源。數據表明TEX細胞傾向于利用葡萄糖而非乙酸生成細胞核乙酰輔酶A,這與ACSS2表達降低相一致。
4.?ACSS2和ACLY分別在TEFF和TEX細胞中協調乙酸鹽和葡萄糖介導的組蛋白乙酰化作用
為了研究乙酸鹽和葡萄糖如何被利用來乙酰化組蛋白,利用同位素示蹤([1,2-13C] acetate 或 [U-13C] glucose)追蹤乙酰基的流向,并結合質譜、流式/質譜流式分析特定組蛋白乙酰化位點。結果表明TEFF細胞優先利用乙酸衍生的乙酰輔酶A來完成所有檢測到的組蛋白乙酰化位點修飾(圖3F),而TEX細胞則傾向于以葡萄糖為主要來源(圖3G)。此外,在CD8+T細胞中敲除Acss2和Acly顯示,與對照組或缺失Acly的細胞相比,缺乏Acss2的TEXprog細胞基于流式細胞術測量的H3K27ac總量降低了近50%,這強調了TEXprog細胞對ACSS2在維持全局組蛋白乙酰化水平方面的高度依賴性(圖3H)。相比之下,Acly的缺失對TEXprog或TEXterm細胞的全局H3K27ac水平沒有影響。實際上,它在TEXterm細胞中趨向于增加H3K27ac(圖3H),這可能是由于在Acly缺陷細胞中觀察到ACSS2的代償性增加(圖3I)。
此外,來自13C-乙酸鹽的組蛋白乙酰化在sgAcss2?CD8+?T細胞中受損,而在sgAcly細胞中增加(圖3J),這再次與ACSS2代償一致(圖3I)。相反,來自13C-葡萄糖的組蛋白乙酰化在sgAcly或sgAcss2?T細胞之間沒有顯著差異。這些發現表明,隨著CD8+T細胞從TEFF分化為TEX細胞,或更準確地說,從TEXprog向TEXterm細胞發展的過程中,它們對組蛋白乙酰化的依賴從ACSS2轉變為ACLY,因此從乙酸鹽轉變為葡萄糖。然而,TEX細胞無法維持與其功能更強的對應細胞(TEFF和TEXprog細胞)相同的全局組蛋白乙酰化水平。
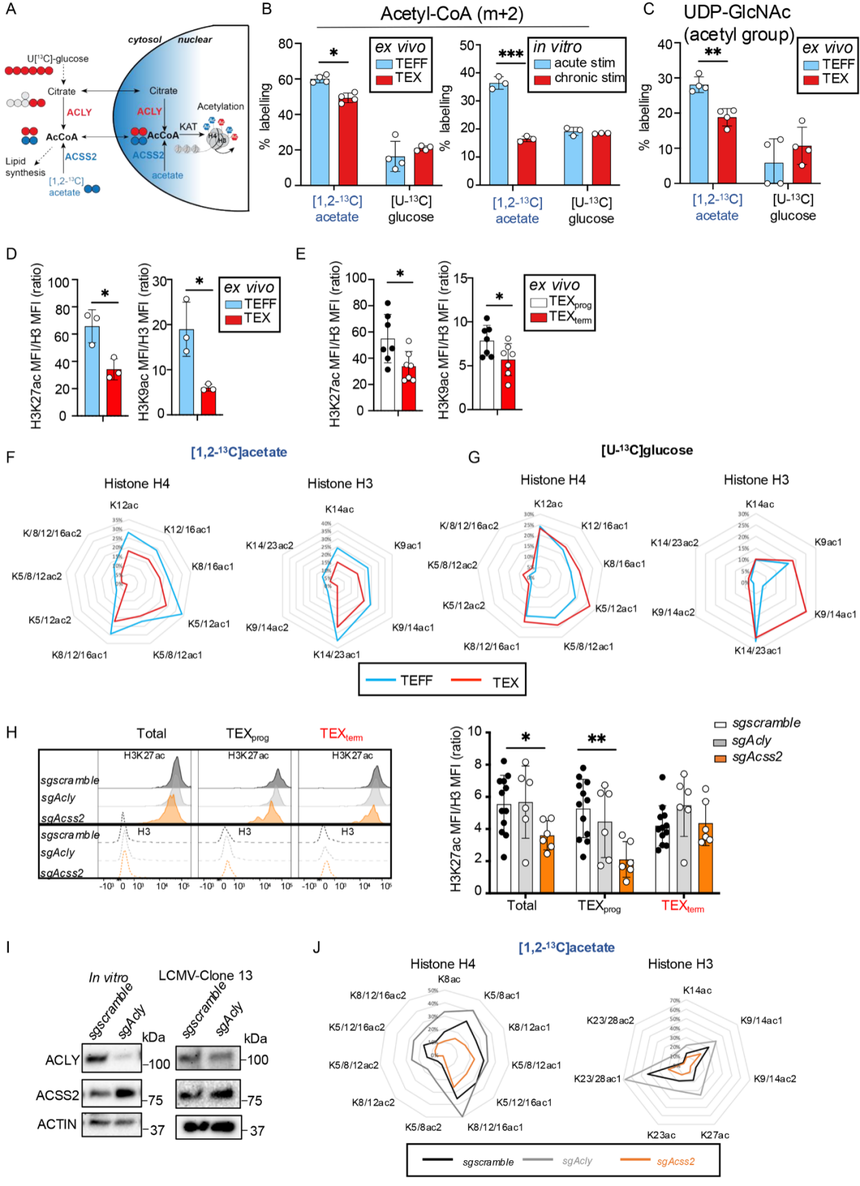
圖3:ACSS2與ACLY在效應T細胞(TEFF)和耗竭T細胞(TEX)中差異調控乙酰輔酶A生成及組蛋白乙酰化修飾。
5.?乙酸鹽衍生的核內乙酰輔酶A促進TEXprog細胞形成
為確定乙酸介導的組蛋白乙酰化和TEXprog細胞分化是否由ACSS2介導的局部核內乙酰輔酶A產生驅動,研究構建了FLAG標記的ACSS2WT、ACSS2NLS(核定位信號標記)和ACSS2NES(核輸出信號標記)的RV過表達載體,并轉導體外激活的P14+CD8+T細胞。這些構建體在CD8+T細胞中產生了類似水平的ACSS2,且具有預期的細胞定位。乙酸依賴性組蛋白乙酰化在ACSS2NLS過表達的CD8+T細胞中最為顯著,證明了核內ACSS2的直接表觀遺傳效應。研究用這三種ACSS2 RV或空載體(EV,pMIG)對照轉導P14+?sgAcss2?CD8+?T細胞,并將供體細胞轉移到隨后感染LCMV-clone 13的小鼠體內。與EV對照和ACSS2NES過表達細胞相比,ACSS2WT和ACSS2NLS過表達的P14+sgAcss2?CD8+T細胞增加了總體和TEXprog供體P14+細胞的比例以及它們產生IFNγ和TNF的能力,盡管ACSS2NLS過表達的效果更為顯著。綜上所述,這些發現強調了乙酸衍生的核內乙酰輔酶A在組蛋白乙酰化和TEXprog細胞形成及功能中的關鍵作用。
6.?ACSS2與ACLY分別與p300和KAT2A共定位
核內乙酰輔酶A通過組蛋白乙酰轉移酶(HAT)的活性來修飾組蛋白。鑒于ACSS2和ACLY對TEX細胞分化和組蛋白乙酰化的影響,作者研究了ACSS2、ACLY與特定HAT之間潛在的協同作用。在P14+CD8?T細胞里,用CRISPR-Cas9把常見HAT基因逐一敲掉,再放入LCMV-clone 13慢性感染小鼠,看缺哪個HAT會改變 TEXprog/TEXterm的比例。結果顯示,sgEp300和sgCrebbp增強了TEXterm細胞的形成,這與sgAcss2的作用相似;而sgKat2a則促進了TEXprog細胞的形成,這與sgAcly的作用類似(圖4A)。這些數據表明,ACSS2與p300(由Ep300編碼)或CBP(由Crebbp編碼)之間,以及ACLY與KAT2A之間存在協同功能。
盡管p300和CBP在結構和功能上密切相關,通常被合稱為p300/CBP,但作者將p300作為ACSS2的功能伙伴進行重點研究,因為在LCMV-克隆13感染期間,與sgCrebbp相比,sgEp300在增強TEXterm細胞和擴增病毒特異性CD8+T細胞方面表現出更強的效果(圖4A)。相應地,p300蛋白在急性刺激的細胞中水平升高,而KAT2A在慢性刺激的細胞中水平上調,這與ACSS2和ACLY的蛋白水平及其各自對TEXprog與TEXterm細胞分化的影響相一致。然后,研究關注了蛋白共定位與相互作用。近鄰連接實驗(PLA)測“距離<40nm的蛋白”會亮,結果顯示在TEFF(急性刺激)里,只看到ACSS2+p300的紅點;在TEX(慢性刺激)里,只看到ACLY+KAT2A的紅點;把ACSS2或ACLY敲掉,紅點消失,證明特異(圖4B和C)。Co-IP+Western blot實驗和熒光顯微鏡也進一步證明內源性ACLY在TEX細胞中與KAT2A相互作用,但不與p300相互作用,而ACSS2在TEFF細胞中特異性地與p300共定位,但不與KAT2A共定位。
研究還進行了功能驗證,在細胞里過表達“帶核定位信號”的ACSS2(ACSS2^NLS),再把p300敲掉,TEXprog細胞積累還是起不來,說明ACSS2必須借p300才有作用;類似地,過表達ACLY^NLS+再敲Kat2a,則TEXprog得以“救回來”,說明ACLY的促TEXterm效應依賴KAT2A。ACSS2^NLS或ACLY^NLS本身都會提高H3K27ac水平,但前提分別是p300或KAT2A存在。
總體而言,這些結果表明ACSS2向p300提供核定位的乙酰輔酶A,促進TEXprog細胞的發育。然而,隨著慢性刺激ACSS2的豐度下降,ACLY提供的乙酰輔酶A支持KAT2A的催化活性占主導地位,促進TEXterm細胞的形成。
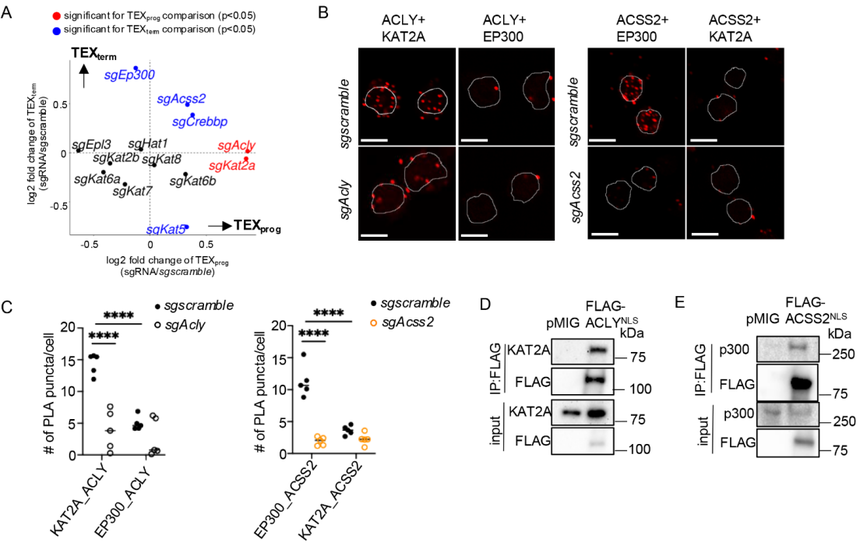
圖4:ACSS2與ACLY分別與p300和KAT2A發生相互作用。
7.?ACSS2與ACLY分別協同p300和KAT2A,共同調控組蛋白乙酰化修飾
基于上面的數據,作如下假設:ACSS2-p300復合物通過控制特定基因位點的組蛋白修飾,來調控與TEXprog細胞相關的基因表達;而ACLY-KAT2A復合物則可能控制著決定TEXterm細胞分化的基因位點的乙酰化。在感染后第21天分選兩類CD8?T細胞,做CUT&Tag測序,分別構建H3K27ac、H3K9ac、H4ac的差異修飾區域(DMR)的全基因組圖譜。將DMR與其鄰近基因進行關聯分析后發現,TEXprog和TEXterm細胞中,各自“特征”基因(例如,TEXprog的Tcf7、Bach2和TEXterm的Havcr2、Il10)的乙酰化水平均有升高。但從全基因組角度看,TEXterm的乙酰化總量低于TEXprog(呼應前文流式檢測)。
研究在P14+CD8+T細胞中敲除了Acss2、Acly、Ep300和Kat2a,并檢查了它們在體外或體內的TEX分化,并對H3K27ac、H3K9ac和H4ac進行了CUT&Tag。如預期,由于慢性刺激的CD8+T細胞中ACSS2自然含量較低,sgAcss2和sgscramble?CD8+T細胞之間觀察到的差異修飾區域(DMR)很少。相比之下,將sgAcly細胞與對照組比較,約4838個DMR在H3K27ac、H3K9ac和H4ac上表現出同步增加,包括許多TEXprog特征位點(如Tcf7、Il7R、Sell、Tnf和Id3),而約8793個DMR在TEXterm特征基因位點(如Il10、Runx3、Havcr2和Entpd1)表現出同步減少(圖5A和B)。
基因集富集分析(GSEA)進一步證實了sgAcly細胞中TEXprog分化的增強(圖5C)。Motif分析顯示,sgAcly T細胞中乙酰化增加的DMR高度富集于AP1-bZIP轉錄因子結合基序(如Fos、Jun、AP-1和ATF),這些與T細胞激活、效應功能以及防止CD8+T細胞耗竭相關,而乙酰化減少的DMR則富集于ETS轉錄因子家族成員(圖5D)。此外,Acly和Kat2a依賴的H3K27ac DMR的全基因組重疊揭示了幾個共同靶向位點,包括許多TEXterm位點。相比之下,sgAcly細胞中上調的DMR(作為Acss2依賴位點的代表)與Ep300依賴位點的重疊顯示了許多TEXprog位點(圖5E)。這進一步強調了ACLY和ACSS2分別以KAT2A和p300依賴的方式調節位點特異性組蛋白乙酰化,這與它們的物理相互作用和功能相互依賴性一致。
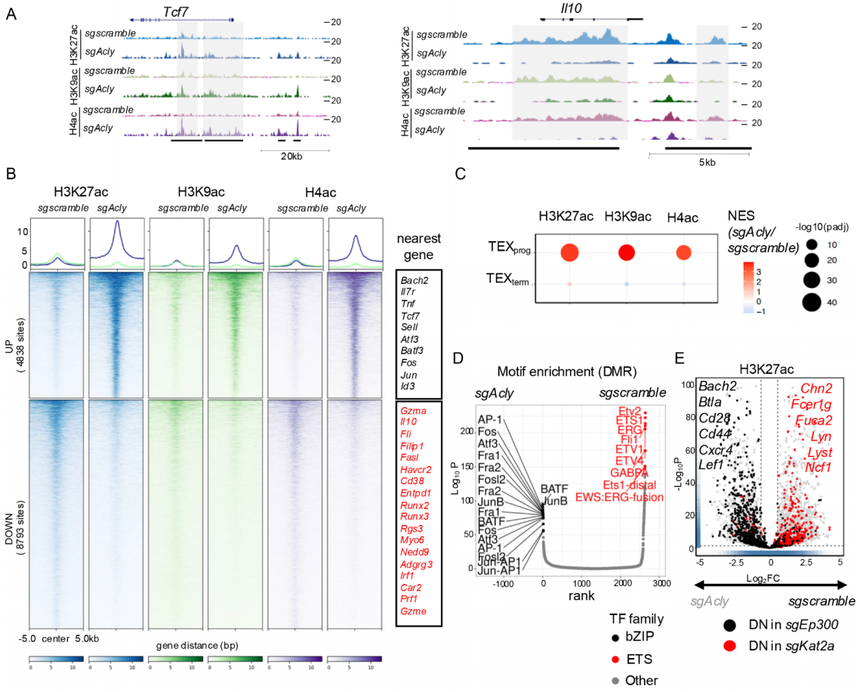
圖5:ACSS2與ACLY分別協同p300和KAT2A,調控位點特異性的組蛋白乙酰化修飾。
8.?核內ACSS2促進TEXprog細胞分化
鑒于過表達ACSS2NLS能夠促進LCMV-clone 13感染期間的組蛋白乙酰化和TEXprog分化,研究者推測這也可能對抗腫瘤免疫產生有益效果。為了驗證這一點,研究在B16-GP33腫瘤植入后第10天將空載體對照或ACSS2NLS過表達的P14+T細胞過繼轉移到小鼠體內(圖6A)。通過流式細胞術檢測,在轉移后第10天,ACSS2NLS過表達的腫瘤浸潤淋巴細胞(TILs)顯示全局H3K27ac和H3K9ac水平增加(圖6B)。此外,對H3K27ac、H3K9ac和H4ac進行的CUT&Tag分析表明,ACSS2NLS過表達的腫瘤浸潤淋巴細胞在TEXprog特征基因(如Tcf7和Bach2)處有特定的組蛋白乙酰化富集(圖6,C和D)。而且,RNA-seq分析顯示ACSS2NLS腫瘤浸潤淋巴細胞中關鍵TEXprog相關基因(Id3、Egr2、Tcf7和Slamf6)表達增加,而關鍵TEXterm相關基因(Id2、Cish和Traf4)表達降低(圖6E)。這些數據證明了核內ACSS2對TILs中TEXprog相關基因表達的影響。即把ACSS2強行送進細胞核(ACSS2^NLS過表達,簡稱OE)不僅能在慢性感染模型里促進TEXprog的形成,還能在腫瘤模型中改善腫瘤浸潤淋巴細胞(TIL)的表觀遺傳狀態和功能。
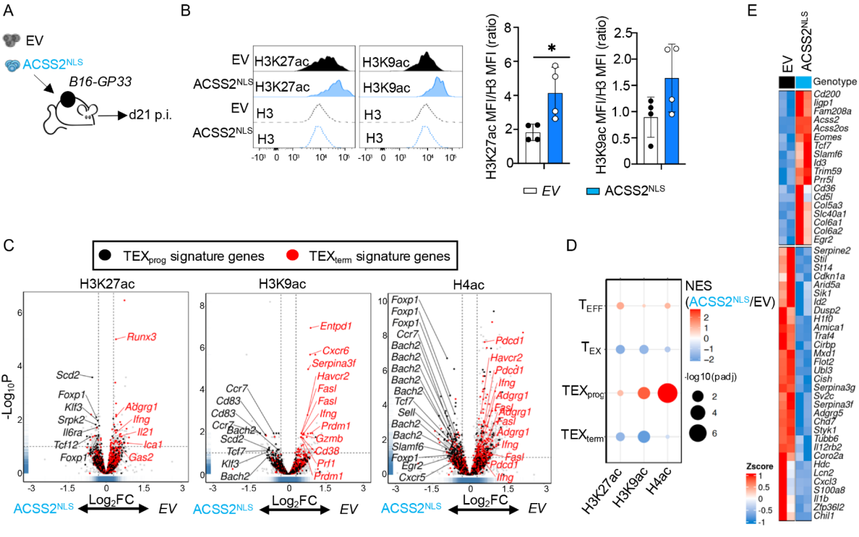
圖6: 細胞核內ACSS2過表達促進TEXprog相關基因座組蛋白乙酰化并增強基因表達。
9.?核內ACSS2過表達和ACLY抑制與ICB協同增強抗腫瘤T細胞應答
研究測試了在腫瘤浸潤淋巴細胞(TIL)中強制表達核內ACSS2是否能增強αPD-L1免疫檢查點阻斷(ICB)的治療效果。結果顯示,與空載體(EV)對照組相比,過表達ACSS2-NLS的TILs無論是在單獨作用還是與ICB聯用時,其祖細胞樣耗竭性T細胞(TEXprog)的形成均得到增強(圖7A),細胞因子產量也更高(圖7B)。它們還會遷移至缺氧區域附近。因此,過表達ACSS2-NLS的TILs能有效控制腫瘤,并與ICB表現出強大的協同作用(圖7C);而使用特異性抑制劑VY-3-135(49)抑制ACSS2則會削弱ICB介導的抗腫瘤免疫應答。
此外,鑒于ACSS2在Acly缺陷型CD8+T細胞中水平升高,研究推斷直接抑制ACLY或許是另一種富集TEXprog細胞并提高ICB療效的方法。的確,使用抑制劑BMS-303141抑制ACLY后,活化的CD8+T細胞中ACSS2的表達顯著增強(圖7D),并在體外耗竭培養中促進了TEXprog細胞的形成。同樣地,在小鼠體內施用BMS-303141也輕微增加了TEXprog的形成,盡管在與ICB聯用組中未觀察到比單獨使用ICB更顯著的效果(圖7E)。然而,BMS-303141與ICB的聯合治療極大地促進了TIL的累積,單獨使用BMS-303141治療也顯示出類似趨勢(圖7F)。相應地,BMS-303141治療能有效抑制腫瘤生長,并以一種依賴于CD8+T細胞的方式與ICB產生協同效應(圖7G)。
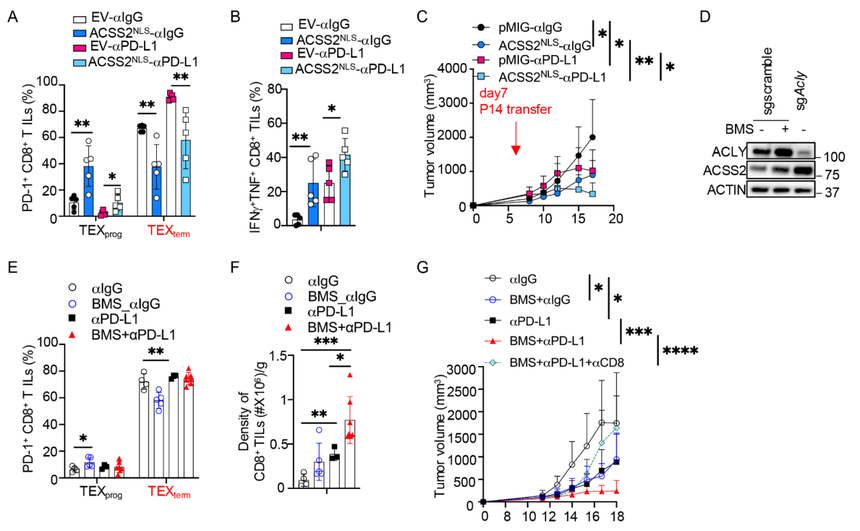
圖7:核內ACSS2過表達或ACLY抑制可增強CD8+T細胞的抗腫瘤免疫功能
? ?研究結論???
該研究揭示了在癌癥和慢性病毒感染中,營養代謝對CD8+T細胞耗竭(TEX)分化的影響機制。研究發現TEX細胞通過下調乙酰輔酶A合成酶2(ACSS2)而維持ATP檸檬酸裂解酶(ACLY)活性,從而將代謝從乙酸轉向檸檬酸。這種代謝轉換通過KAT2A-ACLY相互作用增加了檸檬酸依賴的組蛋白乙酰化,同時通過p300-ACSS2復合物降低了乙酸依賴的組蛋白乙酰化。過表達核定位的ACSS2或抑制ACLY可以防止TEX分化并增強腫瘤特異性T細胞反應。這些發現揭示了一個調控CD8+T細胞分化的營養介導的組蛋白密碼,為基于代謝和表觀遺傳的T細胞治療提供了新思路。
本文提到的相關技術,如有需求歡迎咨詢👏~
{ 往 期 精 彩 回 顧 }
-
項目文章Adv sci(IF:14.1)|ATAC-seq+CUT&Tag表觀多組學助力解析頭頸部鱗狀細胞癌的耐藥性機制
-
化學TOP | 轉錄組測序助力客戶解析枸杞高分支果膠多糖與機械屏障互作調節腸道免疫的分子機制
-
如何研究轉錄因子,合集文章讓你的研究直接開掛
-
如何制備動物組織單細胞懸液,從經驗積累到標準化操作全解析


)
)




)





)



)
