目錄
1.在BAM文件中根據指定的變異等位基因分數的指定位置或區域隨機選擇read。
2.篩選變異等位基因分數的reads:
3.裝BWA和samtools軟件包(samtools在linux系統中下載過,前文有講過)
4.寫py腳本
5.下載pysam庫模塊
6.下載參考基因組hg38
7.解壓gz
8.建立samtools索引
9.建立BWA索引
10.下載picard工具包
11.對hg38.fa建立索引
12.使用BWA的index命令為參考基因組文件創建索引
13.BWA進行比對
14.對bam文件進行標記重復并移除重復的讀取
15.使用變體檢測工具:運行GATK、FreeBayes、Samtools等變體檢測工具,在比對后的數據中檢測SNV和Indel,并生成VCF文件。
16.下載transverse colon的wgs的vcf文件
?編輯
17.尖峰點突變(SNV 和 InDel)進入 bam 文件。(Illumina平臺)
18、變異檢測驗證(FreeBayes / GATK / samtools)
19、Tips 與建議
淺淺介紹一下VarBen是什么東東吧。
VarBen是用于向bam文件添加變異模擬的工具,包括單核苷酸變異、短的插入和缺失以及結構變異。
VarBen主要在linux系統下使用,無需編譯即可運行。但是需要預先配置運行環境。
采用的是比對到參考基因組特定位點的測序reads進行編輯的方式來進行模擬。這種方法可保留測序過程中產生的錯誤類型分布模式,使生成的模擬數據更接近實際數據。那么我們應該怎么做呢?請聽我一本正經的胡說八道吧~
1.在BAM文件中根據指定的變異等位基因分數的指定位置或區域隨機選擇read。
【概念:等位基因分數是一個用于描述在某個基因座中,某個特定等位基因出現的頻率或比例的指標。】
首先,確保你的BAM文件已經被索引。你可以使用samtools index命令為BAM文件創建索引。這個索引文件(.bai)將允許你快速查詢BAM文件中的特定區域。
(具體操作在前文有講過哦~)

利用samtools view命令,你可以提取指定區域或位置的reads(提取染色體chr1上位置10000到20000之間的reads)。
samtools view sorted_ENCFF845HFA.bam chr1:10000-20000 >extracted_reads.bam
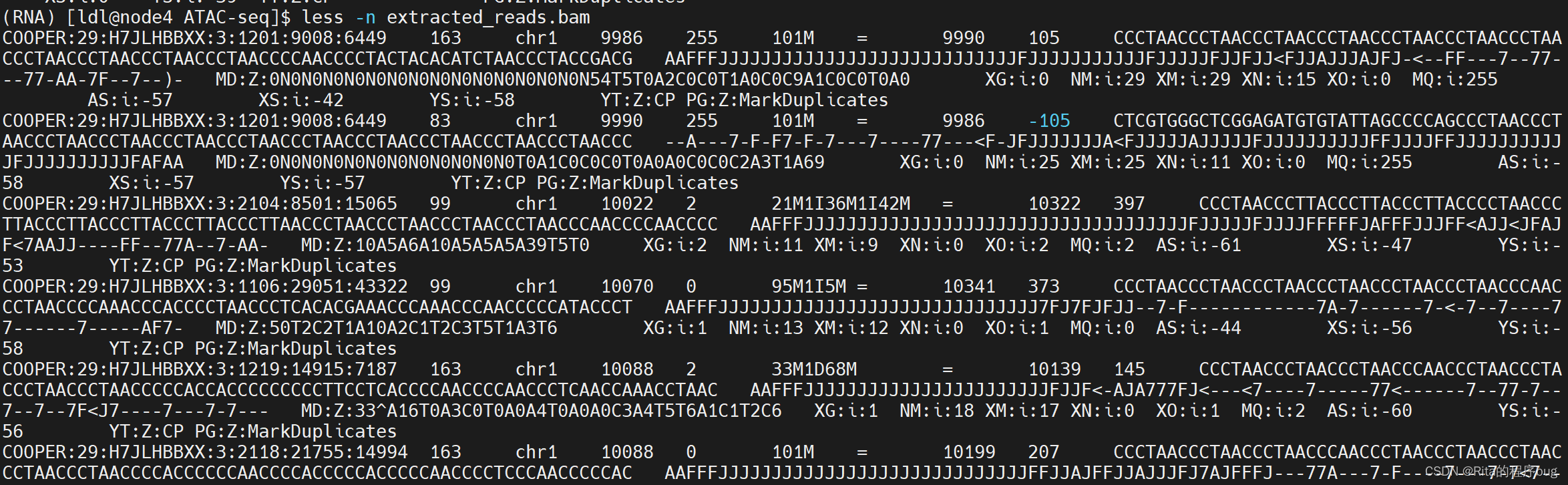
查看文件:
less -n extracted_reads.bam
2.篩選變異等位基因分數的reads:
這一步通常需要使用一些自定義的腳本或工具,因為標準的SAMtools并不直接支持根據等位基因分數篩選reads。你可以使用Python的pysam庫或者其他編程語言的相關庫來讀取BAM文件,并篩選出具有特定等位基因分數的reads。
大家放心,在Linux中使用Python是非常簡單的,因為大多數Linux發行版都預裝了Python。
#檢查python版本
python --version

(我的是python3版本,你的是什么版本呢?)
3.裝BWA和samtools軟件包(samtools在linux系統中下載過,前文有講過)
#如果你也是python3版本的話
pip3 install BWA
#如果不是
pip install BWA下載完成標志:
4.寫py腳本
我在linux專欄中有講到vim文本編輯器,可以用來編輯python腳本的。除此之外,你還可以使用任何文本編輯器(如nano、emacs、gedit等)來編寫Python代碼,并將其保存為.py文件。然后,你可以使用上述方法在終端中運行它。
vim reads.py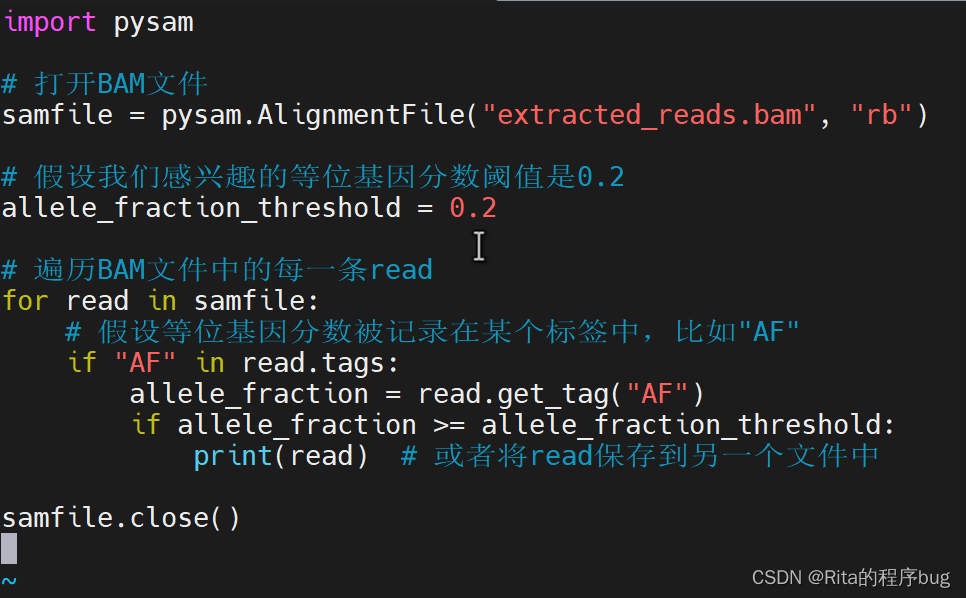
vim命令:
i進入編輯模式
Esc退出編輯
:wq保存并退出
【忘記的話可以回去看看我之前寫的文章(linux專欄)哦】
5.下載pysam庫模塊
pip3 install pysam我一開始以為我以為python3會自帶的,一運行腳本發現報錯,原來是我自作多情了,所以如果你也沒有安裝的話,需要安裝一下呢。(以下為報錯信息)

 本地開發環境搭建指南)








)
)







)