小羅碎碎念
在醫學AI領域,癌癥的精準診斷與預后預測一直是關鍵研究方向。
這篇文章提出了Pathomic Fusion這一創新框架,致力于解決現有方法的局限。

傳統上,癌癥診斷依賴組織學與基因組數據,但組織學分析主觀易變,基因組分析難以精準區分腫瘤與正常細胞特征,且多數深度學習方法僅基于單一數據模態,未充分利用多模態數據互補信息。
文章詳細闡述了Pathomic Fusion框架的構建與實現。
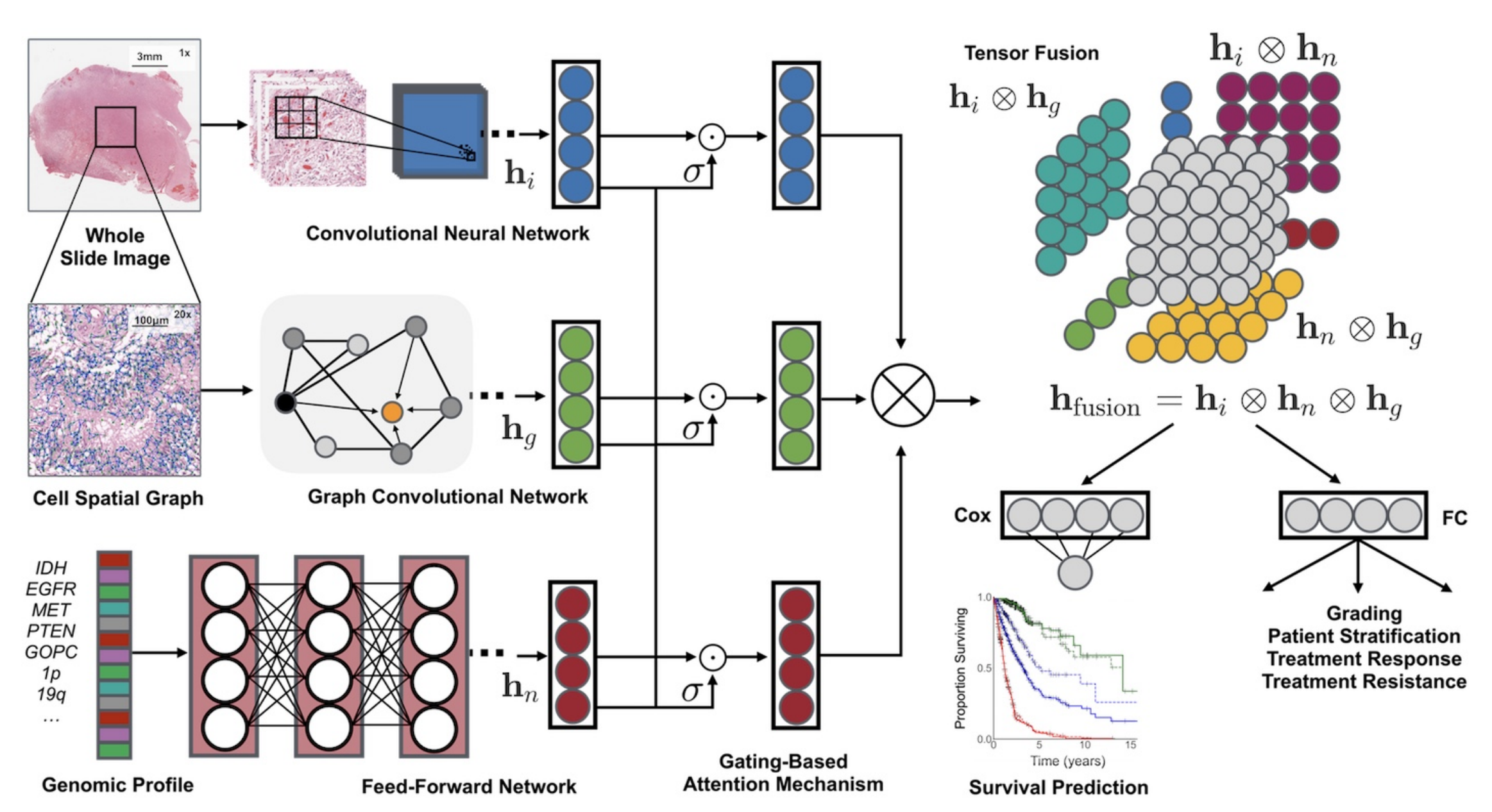
它融合了組織學圖像、細胞圖和基因組特征,利用卷積神經網絡(CNN)和圖卷積神經網絡(GCN)從組織學圖像中提取特征,通過自歸一化網絡處理基因組數據。同時,采用基于門控的注意力機制和克羅內克積來控制特征表達、建模特征交互。
在實驗方面,研究人員利用來自癌癥基因組圖譜(TCGA)的膠質瘤和透明細胞腎細胞癌數據進行15折交叉驗證,結果顯示該框架在生存結果預測和患者分層上優于單模態網絡和傳統分級方法,并且具有良好的可解釋性。
對于從事醫學AI研究的人員來說,這篇文章的價值不容小覷。Pathomic Fusion框架為多模態數據融合提供了新思路,其成功應用證明了整合不同數據模態能提升癌癥預測的準確性和可靠性。此外,文中的方法具有可擴展性,可應用于其他癌癥類型和醫學問題。
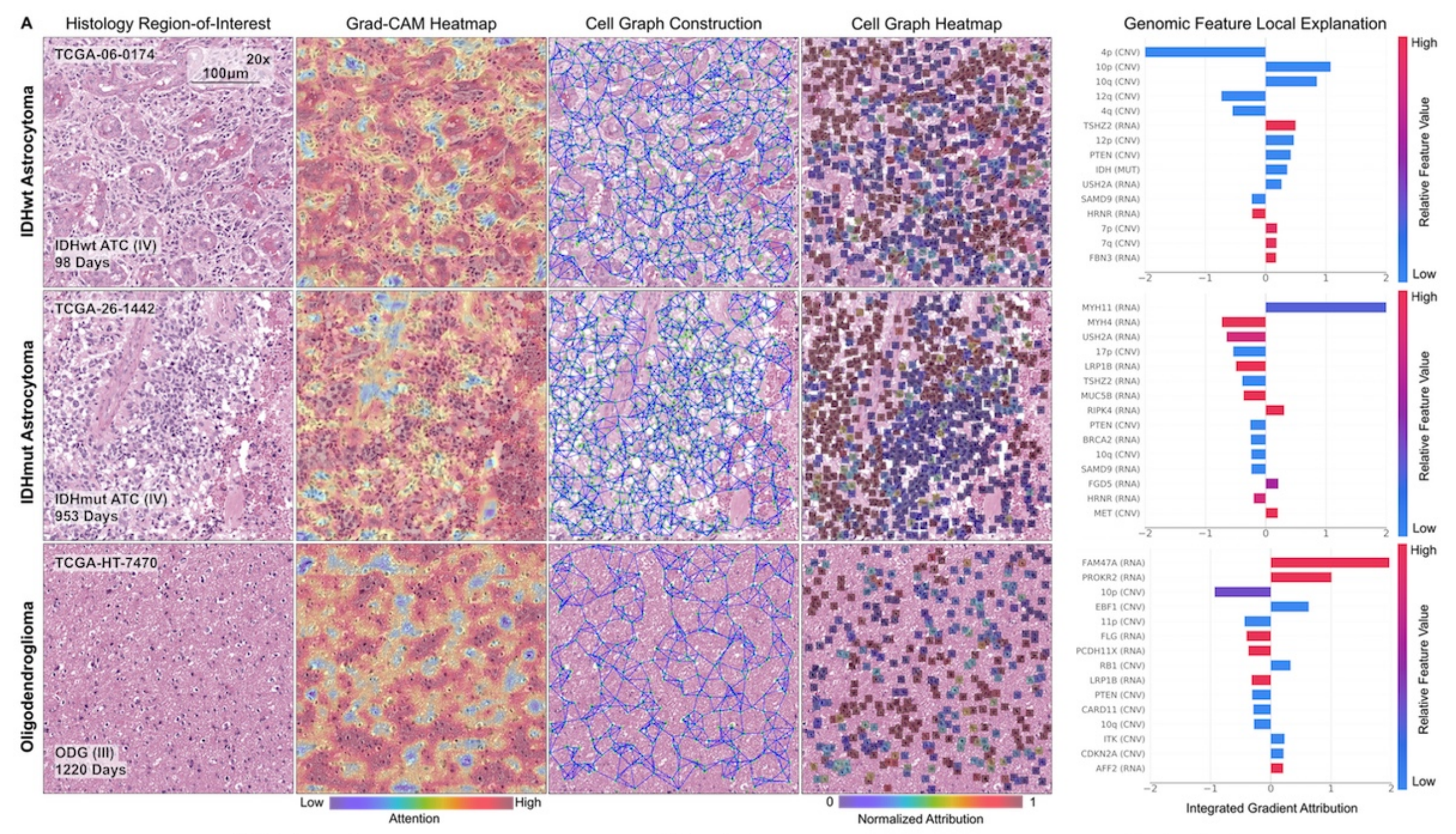
研究中的可解釋性分析方法也為理解模型決策過程提供了有效手段,有助于開發更值得信賴的醫學AI模型,推動醫學AI在癌癥診療領域的進一步發展。
交流群
歡迎大家加入【醫學AI】交流群,本群設立的初衷是提供交流平臺,方便大家后續課題合作。
目前小羅全平臺關注量61,000+,交流群總成員1400+,大部分來自國內外頂尖院校/醫院,期待您的加入!!
由于近期入群推銷人員較多,已開啟入群驗證,掃碼添加我的聯系方式,備注姓名-單位-科室/專業,即可邀您入群。
知識星球
對推文中的內容感興趣,想深入探討?在處理項目時遇到了問題,無人商量?加入小羅的知識星球,尋找科研道路上的伙伴吧!
一、文獻概述
文章提出Pathomic Fusion框架,融合組織學和基因組特征,通過實驗驗證其在癌癥診斷和預后預測方面的優勢,為多模態生物醫學數據的深度學習提供了新方法。
- 研究背景:癌癥診斷、預后和治療反應預測依賴組織學和基因組數據,但現有方法存在局限性。組織學分析主觀且存在觀察者間差異,基因組分析無法精準區分腫瘤與非腫瘤細胞的基因變化。多模態深度學習雖發展迅速,但生物醫學領域的融合策略尚待探索。
- 研究方法
- Pathomic Fusion框架:創新性地融合組織學圖像、細胞圖和基因組特征。組織學特征通過CNN和GCN提取,基因組特征由自歸一化網絡提取,采用基于門控的注意力機制和克羅內克積構建多模態張量,實現特征交互建模。
- 實驗設計:收集TCGA中膠質瘤和透明細胞腎細胞癌數據,進行15折交叉驗證。對比不同模型配置和融合策略,以Cox比例風險模型為基線,使用一致性指數(c-Index)等指標評估模型性能。
- 實驗結果
- 生存預測性能:Pathomic Fusion在膠質瘤和透明細胞腎細胞癌的生存預測上優于單模態網絡和WHO分級范式。在膠質瘤中,c-Index達到0.826,相比WHO范式和之前的方法有顯著提升。
- 患者分層能力:能更精細地分層患者生存曲線。在膠質瘤中,其數字分級與分子亞型相關,可更好地區分中高風險患者;在透明細胞腎細胞癌中,能區分不同生存時長的患者,且分級與Fuhrman分級系統相符。
- 可解釋性:通過修改Grad-CAM和Integrated Gradients方法,可解釋模型在生存預測中對各模態特征的使用。在膠質瘤和透明細胞腎細胞癌中,均能識別重要的基因組特征和組織學特征。
- 研究結論:Pathomic Fusion是一種有效的多模態融合框架,可用于構建客觀的圖像組學檢測方法,實現癌癥的精準診斷和預后預測。該方法具有可擴展性和可解釋性,有助于發現新的生物標志物,為癌癥治療提供指導 。
二、模型架構
2-1:多模態融合框架
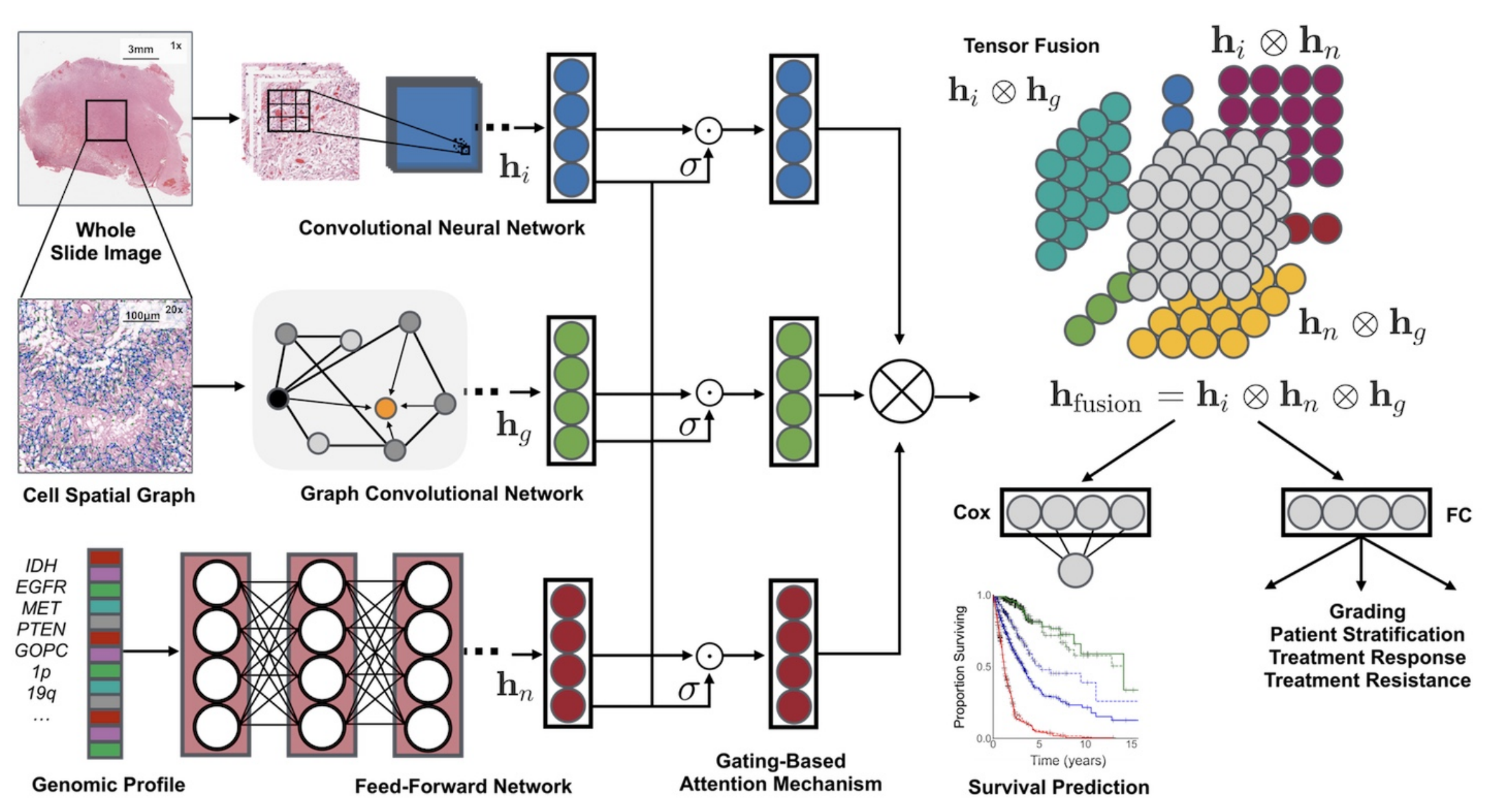
特征提取
- 組織學圖像特征( h i h_i hi?):從全切片圖像(Whole Slide Image)選取區域,經卷積神經網絡(Convolutional Neural Network, CNN)處理提取特征。可單獨用CNN,或結合參數高效的圖卷積網絡(Graph Convolutional Network, GCN) 。
- 細胞空間圖特征( h g h_g hg?):對組織學圖像構建細胞空間圖(Cell Spatial Graph),通過圖卷積網絡(GCN)提取細胞形態等特征。
- 基因組特征( h n h_n hn?):針對基因組圖譜(Genomic Profile ,如IDH、EGFR等基因標識 ),利用前饋網絡(Feed - Forward Network)提取特征。
特征處理與融合
- 門控注意力機制(Gating - Based Attention Mechanism):對各模態特征分別處理,通過門控機制( σ \sigma σ ,類似激活函數)控制各模態特征表達程度,突出重要特征 。
- 張量融合(Tensor Fusion):運用克羅內克積( ? \otimes ? )對處理后的不同模態特征進行兩兩交互建模,得到融合特征 h f u s i o n = h i ? h n ? h g h_{fusion}=h_i \otimes h_n \otimes h_g hfusion?=hi??hn??hg? 。
下游任務應用
- 生存預測(Survival Prediction):將融合特征輸入Cox層,依據Cox比例風險模型預測患者生存情況,輸出生存曲線。
- 分級及其他應用:經全連接層(FC)進行分級,用于患者分層、治療反應預測、治療抵抗評估等。
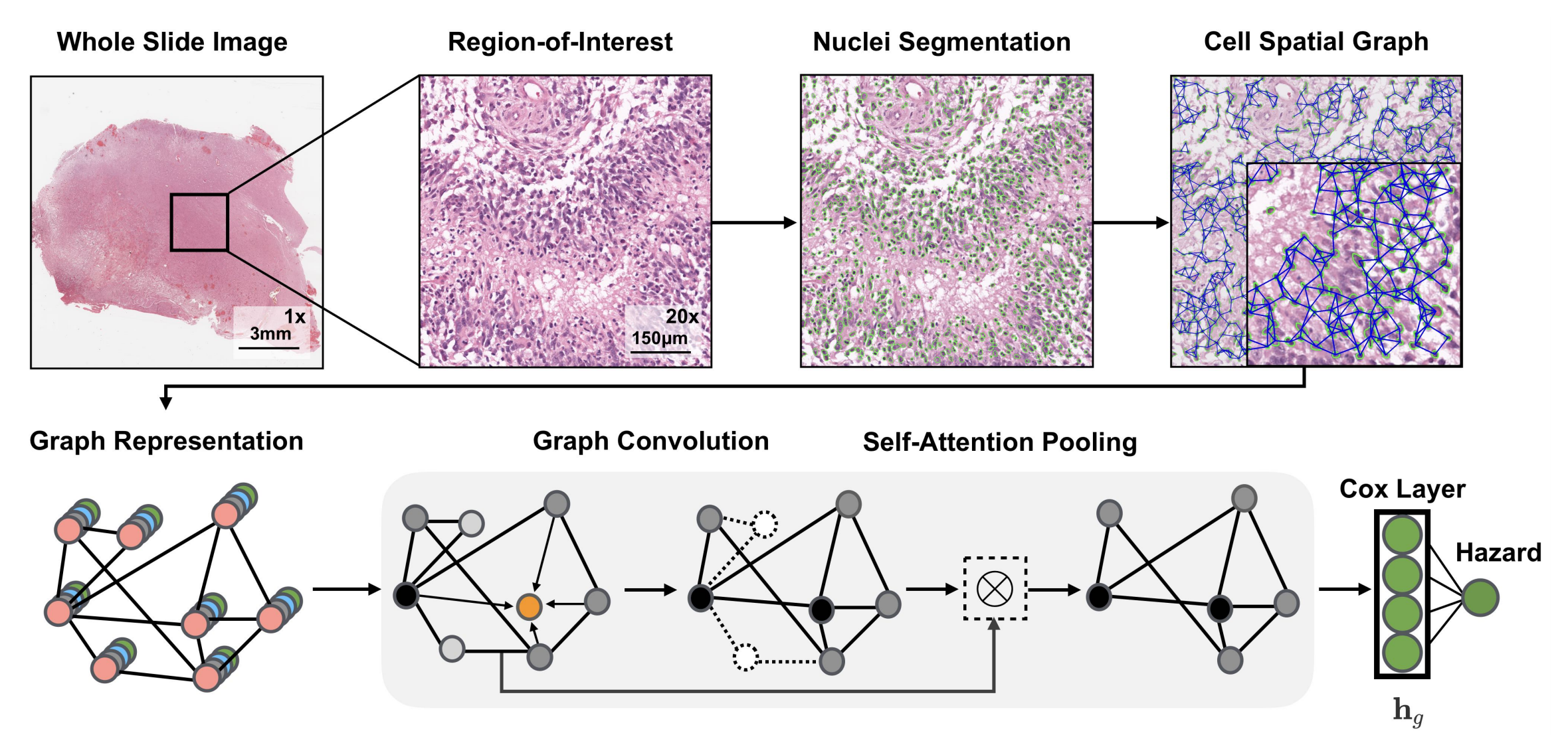
2-2:圖卷積網絡架構
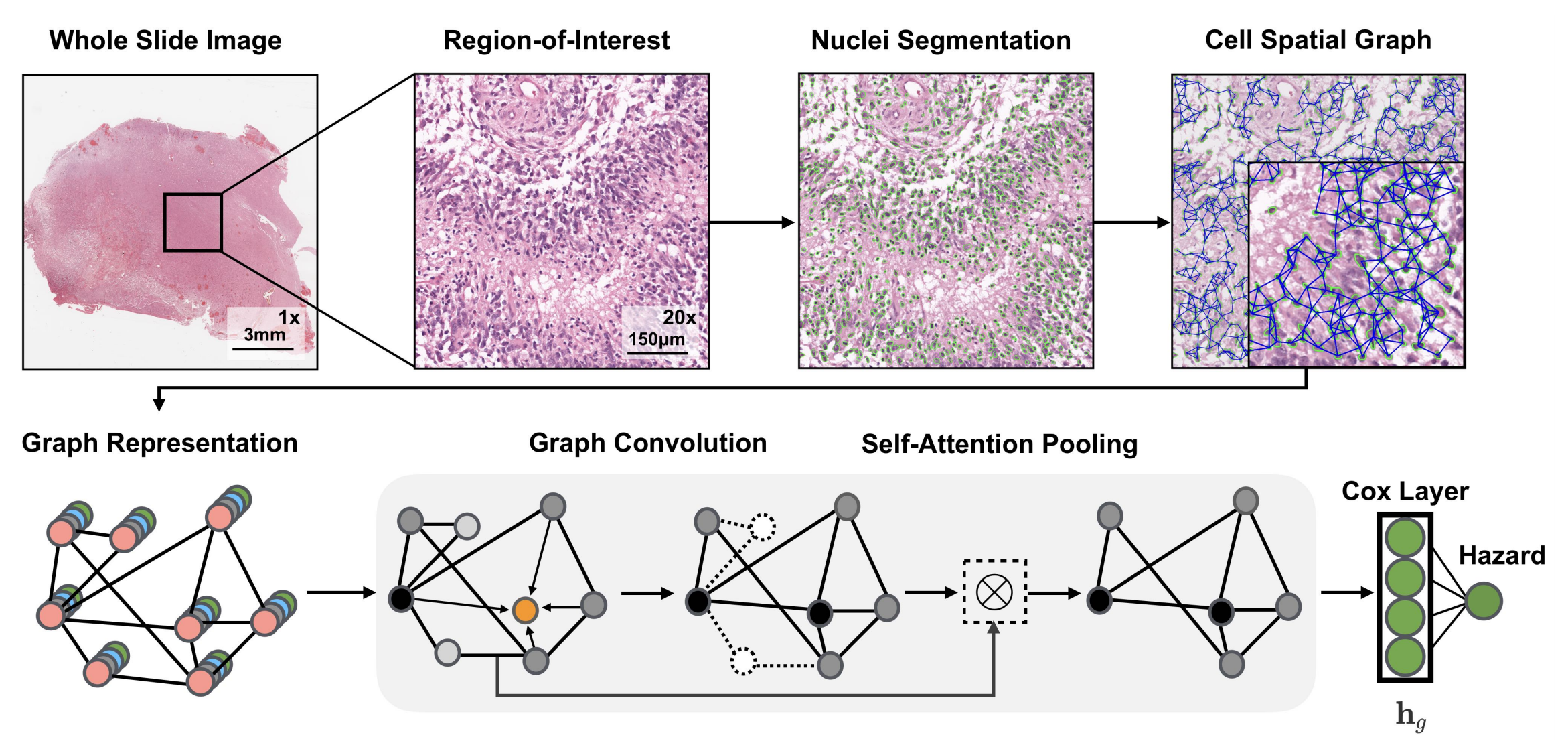
- 全切片圖像(Whole Slide Image)處理:獲取全切片組織學圖像,選取其中感興趣區域(Region - of - Interest ) ,對細胞進行分割(Nuclei Segmentation),構建細胞空間圖(Cell Spatial Graph) ,將細胞視為圖中的節點。
- 圖表示(Graph Representation):基于深度學習的細胞核分割算法分離細胞,用K近鄰(KNN)確定細胞間連接 。細胞特征通過手工設計特征和對比預測編碼學習的深度特征初始化。
- 圖卷積(Graph Convolution):采用GraphSAGE架構的聚合和組合函數,對圖節點特征進行卷積操作,更新節點特征。
- 自注意力池化(Self - Attention Pooling):借鑒SAGEPool的節點掩碼和分層池化策略,通過自注意力機制對圖節點進行池化操作,突出重要節點 。
- Cox層(Cox Layer):將處理后的特征輸入Cox層,計算風險值(Hazard) ,用于生存分析等任務。
三、生存分析
生存分析是對某一事件發生時間進行建模的任務。
在實際情況中,并非所有事件的結果都能完整觀測到,這種情況被稱為刪失(censored)。
以癌癥生存結果預測為例,患者死亡是未刪失事件,而患者存活或最后一次已知隨訪則屬于刪失事件,此時最后一次已知接觸日期就作為生存時間的下限。
設 T T T 為連續隨機變量,表示患者的生存時間,生存函數 S ( t ) = P ( T ≥ t 0 ) S(t)=P(T \geq t_0) S(t)=P(T≥t0?),即表示患者生存時間超過 t 0 t_0 t0? 的概率。
3-1:風險函數
風險函數 λ ( t ) \lambda(t) λ(t) 定義為在 t t t 時刻( t 0 t_0 t0? 之后)事件瞬間發生的概率,其數學表達式為:
λ ( t ) = lim ? Δ t → 0 P ( t ≤ T ≤ t + Δ t ∣ T ≥ t ) Δ t \lambda(t)=\lim_{\Delta t \to 0} \frac{P(t \leq T \leq t + \Delta t | T \geq t)}{\Delta t} λ(t)=Δt→0lim?ΔtP(t≤T≤t+Δt∣T≥t)?
這個公式的含義是,在已經存活到 t t t 時刻的條件下,在極短時間間隔 Δ t \Delta t Δt 內事件發生的概率與 Δ t \Delta t Δt 的比值,當 Δ t \Delta t Δt 趨于 0 時的極限 。
生存函數 S ( t ) S(t) S(t) 與風險函數 λ ( t ) \lambda(t) λ(t) 存在如下關系:
S ( t ) = exp ? ( ? ∫ 0 t λ ( x ) d x ) S(t)=\exp\left(-\int_{0}^{t} \lambda(x)dx\right) S(t)=exp(?∫0t?λ(x)dx)
推導過程
從風險函數定義出發,對 S ( t ) S(t) S(t) 關于 t t t 求導,根據概率的基本性質有:
d S ( t ) d t = ? λ ( t ) S ( t ) \frac{dS(t)}{dt}=- \lambda(t)S(t) dtdS(t)?=?λ(t)S(t)
這是一個可分離變量的微分方程,將其變形為 d S ( t ) S ( t ) = ? λ ( t ) d t \frac{dS(t)}{S(t)}=-\lambda(t)dt S(t)dS(t)?=?λ(t)dt ,然后對兩邊同時積分:
∫ S ( 0 ) S ( t ) d S S = ? ∫ 0 t λ ( x ) d x \int_{S(0)}^{S(t)} \frac{dS}{S}=-\int_{0}^{t} \lambda(x)dx ∫S(0)S(t)?SdS?=?∫0t?λ(x)dx
已知 S ( 0 ) = 1 S(0) = 1 S(0)=1 ,對左邊積分可得 ln ? S ( t ) ? ln ? 1 = ln ? S ( t ) \ln S(t)-\ln 1 = \ln S(t) lnS(t)?ln1=lnS(t) ,所以得到 S ( t ) = exp ? ( ? ∫ 0 t λ ( x ) d x ) S(t)=\exp\left(-\int_{0}^{t} \lambda(x)dx\right) S(t)=exp(?∫0t?λ(x)dx) 。
3-2:Cox比例風險模型
Cox比例風險模型是估計風險函數最常用的半參數方法。
它假設風險函數可以參數化為指數線性函數:
λ ( t ∣ x ) = λ 0 ( t ) e β x \lambda(t|x)=\lambda_0(t)e^{\beta x} λ(t∣x)=λ0?(t)eβx
其中, λ 0 ( t ) \lambda_0(t) λ0?(t) 是基線風險函數,描述了事件在時間 t t t 上的基礎風險,它不依賴于協變量 x x x ; β \beta β 是模型參數向量, x x x 是患者的協變量(如基因特征、臨床指標等), β x \beta x βx 描述了風險如何隨著協變量 x x x 的變化而變化 。
在原始模型中,基線風險 λ 0 ( t ) \lambda_0(t) λ0?(t) 的具體形式未被指定,這使得直接估計 β \beta β 變得困難。不過,可以通過推導Cox部分對數似然函數來估計 β \beta β 。
Cox部分對數似然函數 l ( β , X ) l(\beta, X) l(β,X) 為:
l ( β , X ) = ? ∑ i ∈ U ( X i β ? log ? ∑ j ∈ R i e X j β ) l(\beta, X)=-\sum_{i \in U} \left( X_i \beta - \log \sum_{j \in R_i} e^{X_j \beta} \right) l(β,X)=?i∈U∑? ?Xi?β?logj∈Ri?∑?eXj?β ?
其中, U U U 是未刪失患者的集合, R i R_i Ri? 是死亡時間或最后隨訪時間晚于患者 i i i 的患者集合 。
對 l ( β , X ) l(\beta, X) l(β,X) 關于 β \beta β 求偏導:
? l ( β , X ) ? β = ∑ i ∈ U ( X i ? ∑ j ∈ R i X j e X j β ∑ j ∈ R i e X j β ) \frac{\partial l(\beta, X)}{\partial \beta}=\sum_{i \in U} \left( X_i - \frac{\sum_{j \in R_i} X_j e^{X_j \beta}}{\sum_{j \in R_i} e^{X_j \beta}} \right) ?β?l(β,X)?=i∈U∑?(Xi??∑j∈Ri??eXj?β∑j∈Ri??Xj?eXj?β?)
為了更方便地表示,令 δ ( i ) \delta(i) δ(i) 為指示變量,當患者 i i i 未刪失時 δ ( i ) = 1 \delta(i) = 1 δ(i)=1 ,刪失時 δ ( i ) = 0 \delta(i) = 0 δ(i)=0 ,則偏導可寫成:
? l ( β , X ) ? β = ∑ i ∈ U ( δ ( i ) X i ? ∑ j ∈ R i δ ( j ) X j e X j β ∑ j ∈ R i e X j β ) \frac{\partial l(\beta, X)}{\partial \beta}=\sum_{i \in U} \left( \delta(i)X_i - \frac{\sum_{j \in R_i} \delta(j)X_j e^{X_j \beta}}{\sum_{j \in R_i} e^{X_j \beta}} \right) ?β?l(β,X)?=i∈U∑?(δ(i)Xi??∑j∈Ri??eXj?β∑j∈Ri??δ(j)Xj?eXj?β?)
可以使用迭代優化算法,如牛頓 - 拉夫森(Newton - Raphson)算法或隨機梯度下降(Stochastic Gradient Descent)算法來估計 β \beta β 。
3-3:深度學習在生存分析中的應用及模型評估
訓練用于生存分析的深度網絡時,隱藏層特征作為Cox模型協變量,部分對數似然函數導數作為反向傳播誤差 。
評估生存分析網絡性能用一致性指數(c - Index) ,衡量預測風險得分與患者真實生存時間排序一致性 。為展示Pathomic Fusion性能,與其他模型對比用c - Index 。
臨床實踐基線是在Cox比例風險模型中用真實分子亞型作為協變量 。通過Log Rank檢驗計算P值,評估不同風險分層 ,如膠質瘤的低、中、高風險(33 - 66 - 100百分位數),透明細胞腎細胞癌(CCRCC)的25 - 50 - 75 - 100百分位數風險分層 。
四、方法細節
4-1:基因組和轉錄組特征的納入標準
在對合并的TCGA - GBMLGG和TCGA - KIRC項目進行分析時,分別使用了320個和357個基因組特征。
基因組特征包括突變(如IDH1基因的突變狀態二元指示,0/1 )和拷貝數變異(CNV )(如基因和染色體區域的擴增/缺失拷貝 )。TCGA中拷貝數變異的測量使用Affymetrix SNP 6.0芯片來識別基因組區域的重復拷貝,最終輸出為片段平均值(擴增區域為正值,缺失區域為負值 )。
對于TCGA - GBMLGG,本分析中使用的突變和CNV數據是從Mobadersany等人[29]使用的同一組基因組特征中整理而來。整理的基因包括EGFR、MDM4、MGMT、MYC和BRAF,這些基因與血管生成、細胞凋亡、細胞生長和分化等致癌過程有關。
對于TCGA - KIRC,使用了擴增/缺失最多的基因(所有擴增或缺失大于7%的CNV ),得到117個CNV特征。對于這兩個項目,均納入了RNA - Seq表達數據,其以mRNA轉錄本的定量總體豐度來衡量。
通過cBioPortal,為兩個項目均選擇了前240個差異表達基因[65]。由于基因組特征之間不存在任何明確的空間或時間依賴性,因此直接輸入特征。
4-2:TCGA - GBMLGG中的數據缺失和對齊
為與先前最先進的研究進行比較,使用了[29]附錄中現有的整理后的TCGA - GBMLGG數據,這需要仔細處理多模態數據中的缺失值。
對于每位患者,使用來自診斷切片的1 - 3個20倍放大、1024 × 1024大小(0.5 μ /像素 )的組織學感興趣區域(ROI ),以及320個基因組特征。在769名患者中,72名患者缺失分子亞型(IDH突變和1p19q共缺失 )信息,33名患者缺失組織學亞型和分級標簽,256名患者缺失mRNA - Seq數據(圖6 )。
由于部分患者有多個來自診斷切片的ROI,在交叉驗證中每個圖像被視為一個單獨的數據點,同時復制基因組和真實標簽信息。使用與[29]附錄相同的訓練 - 測試劃分進行15折蒙特卡羅交叉驗證,該劃分按TCGA ID隨機生成,80%用于訓練,20%用于測試。
由于數據缺失,根據任務(生存預測與分級分類 )和使用的模態組合(組織學、基因組、組織學 + 基因組 ),從訓練劃分中選取不同子集來訓練單模態和多模態網絡。
在對存在缺失數據的交叉驗證測試劃分上驗證模型時,對測試劃分進行標準化處理,以排除所有模型中的缺失數據(圖5中的中心重疊部分 )。
在處理透明細胞腎細胞癌(CCRCC )數據時,數據缺失不是問題,所有單模態和多模態網絡均使用相同的訓練 - 測試劃分進行15折交叉驗證。
4-3:網絡架構
本研究使用三種不同的網絡架構來處理三種模態的數據:
- 1)用于組織學圖像的帶批量歸一化的VGG19卷積神經網絡(CNN );
- 2)用于細胞空間圖的圖卷積網絡(GCN );
- 3)用于分子特征譜的前饋自歸一化網絡。
VGG19網絡由16個卷積層、3個全連接層和5個最大池化層組成,輸入圖像大小為512 × 512。在前兩個全連接層(大小為1024 )后應用丟棄率為0.25的Dropout,在最后一個隱藏層(大小為32 )后應用較低丟棄率(p = 0.05 )的Dropout。
GCN由3個GraphSAGE層和自注意力池化層組成,隱藏層維度為128,隨后是兩個大小分別為128和32的線性層。
基因組自歸一化網絡(Genomic SNN )由4個連續的全連接層模塊組成,維度分別為[64, 48, 32, 32],采用ELU激活函數和Alpha Dropout。
對于生存結果預測,所有網絡使用Sigmoid函數激活,輸出縮放至 - 3到3之間。對于分級分類,所有網絡使用Log Softmax激活,以計算三個WHO分級各自的得分。
多模態網絡架構由兩個部分組成:
- 1)基于門控的模態注意力;
- 2)通過克羅內克積進行融合。
每種模態通過三個線性層進行門控,第二個線性層用于計算注意力分數。
對于生存結果預測,使用基因組模態對圖像和圖模態進行門控;對于分級分類,使用組織學圖像模態對基因組和圖模態進行門控。對門控后的單模態特征表示進行額外降維,以減小三模態網絡中克羅內克積特征空間的輸出大小。
在三模態網絡中,對于生存結果預測,基因組模態的第一和第三個線性層有32個隱藏單元以保持特征圖維度,圖像和圖模態的線性層有16個隱藏單元,以便將特征表示轉換為較低維度。
對于分級分類,保持組織學圖像模態的特征維度,降低圖和基因組模態的維度。在任何任務的雙模態網絡中均不進行特征維度降低。
對于特征融合,計算每種模態各自單模態特征表示的克羅內克積,對于CNN?SNN、GCN?SNN和CNN?GCN?SNN,分別創建大小為[33 × 33]、[33 × 33]、[33 × 17 × 17]的特征圖。
為使用未擾動的單模態特征,在計算克羅內克積之前向每個特征向量添加1。在門控和計算多模態張量后插入丟棄率為(p = 0.25 )的Dropout層。
4-4:實驗細節
Pathomic Fusion使用PyTorch 1.5.0、PyTorch Geometric 1.5.0、Captum 0.2.0和Lifelines 0.24.6構建。
用于構建細胞圖的節點特征通過以下方式計算:
- 1)分割每個細胞核;
- 2)使用OpenCV 4.2.0中的輪廓特征工具箱;
- 3)紋理特征工具箱;
- 4)使用對比預測編碼的自監督深度特征;
- 5)使用PyFlann 1.6.14進行圖構建。
實驗使用的資源包括本地工作站上的12塊英偉達GeForce RTX 2080 Ti顯卡,以及谷歌云平臺上的2塊英偉達Tesla V100顯卡。
組織學CNN使用來自ImageNet的預訓練權重進行初始化,隨后使用0.0005的低學習率和8的批量大小對網絡進行微調。通過隨機裁剪512 × 512大小圖像、顏色抖動以及隨機垂直和水平翻轉進行數據增強。
組織學GCN和基因組SNN使用Klambeur等人[58]提出的自歸一化權重進行初始化,分別使用0.002的學習率,以及32和64的批量大小進行訓練。對于基因組SNN,還使用了超參數值為3e - 4的輕度L1正則化以強制特征稀疏性。所有網絡使用Adam優化器、丟棄率p = 0.25以及線性衰減學習率調度器,訓練輪次相同。
在訓練組織學CNN后,對于每個1024 × 1024的組織學ROI,從9個重疊的512 × 512圖像塊中提取[32 × 1]的嵌入向量,將其與各自的細胞圖和基因組特征輸入配對,作為Pathomic Fusion的輸入。
對于組織學GCN和基因組SNN,首先按照上述訓練細節訓練各自的單模態網絡,然后在凍結單模態網絡模塊的情況下,使用0.0001的學習率和Adam求解器訓練多模態網絡的最后線性層。
在第5輪訓練時,解凍基因組和圖網絡,然后使用0.0001的學習率、Adam求解器和線性衰減學習率調度器再訓練網絡25輪。
4-5:評估細節
在15折交叉驗證的測試劃分上評估每個單模態和多模態網絡預測的風險值和分級得分。
為了在TCGA - GBMLGG上基于CNN進行生存結果預測時使用整個1024 × 1024的組織學圖像,與先前工作類似,計算屬于每位患者的所有組織學ROI中9個重疊的512 × 512圖像裁剪塊的風險預測平均值。
為繪制卡普蘭 - 邁耶(Kaplan - Meier)曲線,匯集15折交叉驗證中所有測試劃分的預測風險值,并根據生存時間進行繪制。為創建散點圖(Swarm plots ),在匯集之前對每個劃分中的預測風險值進行z - 分數標準化,以便在可視化中低風險與中風險的得分范圍相似。
對于TCGA - GBMLGG上的分級分類,使用重疊的512 × 512圖像塊的最大softmax激活得分來確定類別。對于在CCRCC上基于CNN的生存結果預測,類似地計算每位患者512 × 512組織學ROI的風險預測平均值。
五、項目梳理
注意,這只是初步梳理,并不是詳細的復現的教程。
Pathomic Fusion 是一個整合組織病理學圖像和基因組特征的多模態融合框架,用于癌癥診斷和預后預測。其核心創新點在于使用注意力門控(Attention Gating)和Tensor融合技術,支持卷積神經網絡(CNN)、圖卷積網絡(GCN)或其組合處理數據。
5-1:環境配置
系統要求
? 操作系統:Linux(推薦 Ubuntu 18.04+)
? 硬件:NVIDIA GPU(如 RTX 2080 Ti 或 V100)
? 依賴項:
? CUDA 10.1 + cuDNN 7.5
? PyTorch ≥1.1.0
? torch_geometric=1.3.0
安裝步驟
# 安裝 PyTorch
conda install pytorch==1.7.1 torchvision==0.8.2 cudatoolkit=10.1 -c pytorch# 安裝 torch-geometric
pip install torch-scatter -f https://pytorch-geometric.com/whl/torch-1.7.0+cu101.html
pip install torch-sparse -f https://pytorch-geometric.com/whl/torch-1.7.0+cu101.html
pip install torch-geometric==1.7.0
5-2:數據準備
目錄結構
按以下結構組織數據:
data/
└─ PROJECT_NAME/├─ Images/ # 組織病理圖像(.png)├─ Graphs/ # 圖數據(.pkl)└─ Genomics/ # 基因組數據(.csv)
數據對齊
? 運行 make_splits.py 生成跨模態數據對齊文件:
python make_splits.py --project PROJECT_NAME --data_root ./data/
? 生成 splits.pkl 文件記錄數據路徑和劃分。
5-3:代碼結構解析
關鍵文件說明
? train_cv.py: 執行交叉驗證訓練。
? test_cv.py: 在測試集評估模型。
? networks.py: 定義單模態和多模態網絡模型。
? fusion.py: 多模態融合機制實現。
? data_loaders.py: 數據加載器,支持多模態輸入。
5-4:模型訓練與評估
單模態訓練(示例:組織病理圖像)
python train_cv.py \--exp_name survival_prediction \--dataroot ./data/TCGA_GBMLGG/ \--task surv \--mode A \--model_name CNN_A \--batch_size 64 \--lr 0.002 \--gpu_ids 0
? mode A: 指定使用圖像模態。
? task surv: 生存預測任務。
多模態融合訓練(圖像+基因組)
python train_cv.py \--task grad \--mode AB \--model_name Fusion_AB \--fusion_type tensor \--lr 0.001 \--niter_decay 100
? fusion_type tensor: 使用Tensor融合策略。
模型測試
python test_cv.py \--checkpoints_dir ./checkpoints/TCGA_GBMLGG/ \--model Fusion_AB \--phase test
5-5:復現論文結果
下載預處理數據
訪問 Google Drive 下載 TCGA-GBMLGG 和 TCGA-KIRC 數據集。
運行基線模型
python run_cox_baselines.py \--omics_path ./data/genomic_data.csv \--survival_path ./data/survival_labels.csv
結束語
本期推文的內容就到這里啦,如果需要獲取醫學AI領域的最新發展動態,請關注小羅的推送!如需進一步深入研究,獲取相關資料,歡迎加入我的知識星球!





)

)




第五期)






